|
|
|
|
|
Drug Metabolism: Phase I and Phase II Metabolism
|
|
-
-
Lipophilic drug properties that promote passage through biological membranes and facilitate reaching site to drug action inhibit drug excretion.
-
Note: renal excretion of unchanged drug contributes only slightly to elimination, since the unchanged, lipophilic drug is easily reabsorbed through renal tubular membranes.
-
-
Biotransformation of drugs to more hydrophilic molecules is required for elimination from the body
-
Biotransformation reactions produces more polar, hydrophilic, biologically inactive molecules -- that are more readily excreted.
-
Sometimes metabolites retain biological activity and may be toxic.
-
-
Drug biotransformation mechanisms are described as either phase I or phase II reaction types.
-
-
-
Phase I and Phase II Reactions -- Overview
-
-
Parent drug is altered by introducing or exposing a functional group (-OH,-NH2, -SH)
-
Drugs transformed by phase I reactions usually lose pharmacological activity
-
Inactive, prodrugs are converted by phase I reactions to biologically-active metabolites
-
Phase I reaction products may:
-
Be directly excreted in the urine
-
React with endogenous compounds to form water soluble conjugates.
-
-
-
-
-
Parent drug participates in conjugation reactions that:
-
Form covalent linkage between a parent compound functional group and:
-
Glucuronic acid
-
Sulfate
-
Glutathione
-
Amino acids
-
Acetate
-
-
-
-
Conjugates are highly polar, and generally biologically inactive. One exception to this rule is a morphine metabolite, morphine glucuronide which is a more potent analgesic compared to the parent compound. Conjugates tend to be rapidly excreted in the urine.
-
High molecular weight conjugates are more likely excreted in the bile. The conjugate bond may be cleaved by intestinal flora with the parent compound released back to the systemic circulation. This process, "enterohepatic recirculation" results in delayed parent drug elimination and a prolongation of drug effects.
-
-
Principal Organs for Biotransformation
-
The Principal Organ for biotransformation is the liver, although other organs participate in metabolism. These other systems include lungs, skin, kidney, and the gastrointestinal tract.
-
Other metabolizing organs:
-
-
Sequence I could be as follows:
-
(1) Oral administration (isoproterenol (Isuprel), meperidine (Demerol), pentazocine (Talwain), morphine)
-
(2) Drug is absorbed intact by the small intestine.
-
(3) Drug is transported to the liver (portal system) where it might be extensively metabolized by the liver, an example of a first-pass effect.
-
-
Sequence II might be as follows:
-
(1) Oral administration (e.g. clonazepam (Klonopin), chlorpromazine (Thorazine)) and
-
(2) Drug is absorbed intact by the small intestine.
-
(3) Extensive intestinal metabolism might ensue, contributing to overall first-pass effects.
-
-
Issues in bioavailability: Reduced bioavailability might result from several factors including
-
(a) the first pass effect in which the bioavailability of orally administered drugs become so limited that alternative routes of administration must be employed.
-
(b) Intestinal flora might metabolize the drug.
-
(c) The drug itself is unstable in gastric acid; an example of this effect would be penicillin.
-
(d) the drug might be metabolized by digestive enzymes; an example of this effect would be insulin. (d) Finally, the drug might be metabolized by intestinal wall enzymes; sympathomimetic catecholamines represent examples of this effect.
-
First pass effect:
-
Bioavailability of orally administered drugs may be so limited that alternative routes of administration must be used
-
-
Intestinal flora may metabolize drugs
-
Drugs is unstable in gastric acid, for example, penicillin
-
Drug is metabolized by digestive enzymes such as nsulin
-
Drug is metabolized by intestinal wall enzymes such as the sympathomimetic catecholamines
-
-
Correia, M.A., Drug Biotransformation. in Basic and Clinical Pharmacology, (Katzung, B. G., ed) Appleton-Lange, 1998, pp 50-61.
Benet, Leslie Z, Kroetz, Deanna L. and Sheiner, Lewis B The Dynamics of Drug Absorption, Distribution and Elimination. In, Goodman and Gillman's The Pharmacologial Basis of Therapeutics,(Hardman, J.G, Limbird, L.E, Molinoff, P.B., Ruddon, R.W, and Gilman, A.G.,eds) TheMcGraw-Hill Companies, Inc.,1996, pp. 3-27
Mixed function oxidase System (cytochrome 450 System)--Phase I Reactions
-
Microsomes have been used to study mixed function oxidases
-
Drug metabolizing enzymes are located in lipophilic, hepatic endoplasmic reticulum membranes. Smooth endoplasmic reticulum contains those enzymes responsible for drug metabolism.
-
-
-
One molecule oxygen is consumed per substrate molecule
-
One oxygen atom -- appears in the product; the other in the form of water
-
Oxidation-Reduction Process:
-
Two important microsomal enzymes:
-
Cytochrome P450: -- terminal oxidase
-
Multiple forms
-
Named cytochrome P450 because:
-
The reduced (ferrous) form, binds carbon monoxide: -- the resulting complex exhibits of absorption maximum at 450 nm.
-
-
NOTE in the Figure Below the CONVERSION OF RH to ROH representing DRUG OXIDATION
-
-
-
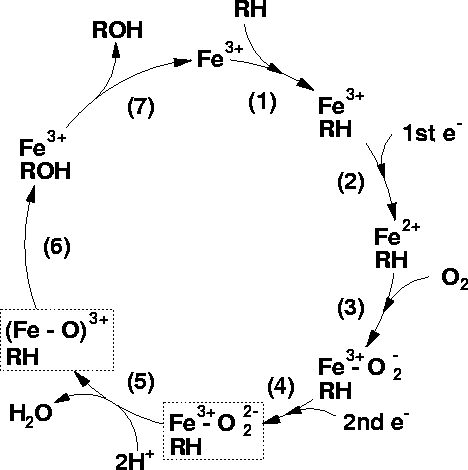
Cytochrome p450 cycle (diagram by Matthew Segall, 1997)
-
"The binding of a substrate to a P450 causes a lowering of the redox potential by approximately 100mV, which makes the transfer of an electron favourable from its redox partner, NADH or NADPH.
-
The first reduction -The next stage in the cycle is the reduction of the Fe3+ ion by an electron transfered from NAD(P)H via an electron transfer chain.
-
Oxygen binding An O2 molecule binds rapidly to the ion Fe2+ forming Fe2+-O2
-
Second reduction A second reduction is required by the stoichiometry of the reaction. This has been determined to be the rate-determining step of the reaction
-
O2 cleavage: The O2 reacts with two protons from the surrounding solvent, breaking the O-O bond, forming water and leaving an Fe-O3+ complex.
-
Product formation The Fe-ligated O atom is transferred to the substrate forming an hydroxylated form of the substrate.
-
Product release The product is released from the active site of the enzyme which returns to its initial state."--Matthew Segall, 1997
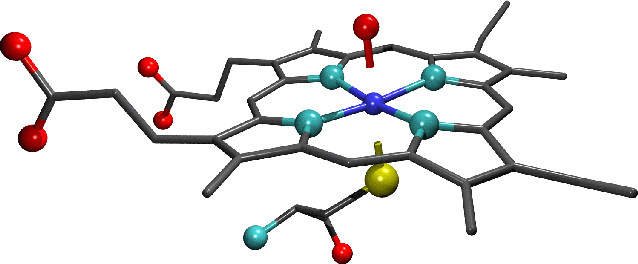
-
"The active site of substrate-free cytochrome p450: Note the water molecule (which can be seen as a single oxygen atom) that forms the sixth axial ligand of the haem iron. Oxygen atoms are shown in red, nitrogen in light blue, sulphur in yellow and iron in dark blue. Carbon atoms are shown in grey as bonds only and hydrogens have been omitted from this figure for clarity."
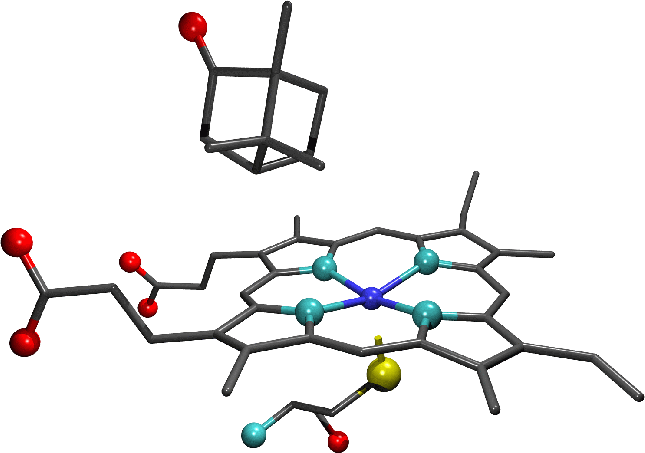
-
"The active site of camphor-bound cytochrome p450cam , an example of a substrate-bound system.
-
Note the absence of the water molecule which formed the sixth axial ligand of the haem iron in the substrate-free enzyme."
-
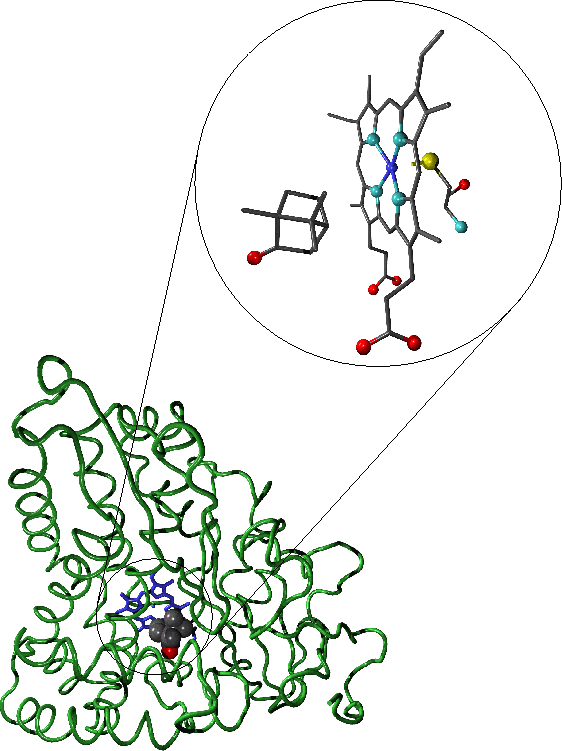
-
" A representation of with bound camphor. The enlarged active site region shows the camphor substrate, haem moiety and cysteine residue which forms the distal haem ligand. In the representation of the full enzyme the protein backbone is shown in green, the haem moiety in blue and the substrate is coloured according to atomic species. Oxygen atoms are shown in red, carbon in grey, nitrogen in light blue, sulphur in yellow and iron in dark blue."-diagrams and text by Matthew Segall, 1997
Stoelting, R.K., "Pharmacokinetics and Pharmacodynamics of Injected and Inhaled Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, 1-17.
Dolin, S. J. "Drugs and pharmacology" in Total Intravenous Anesthesia, pp. 13-35 (Nicholas L. Padfield, ed), Butterworth Heinemann, Oxford, 2000
|
|
|
|
|