|
|
|
Medical Pharmacology Chapter 5: Autonomic Pharmacology: Adrenergic Drugs
|
|
|
|
|
|
|
α- and β-adrenergic receptor selectivity
Adrenergic agonists have been developed which exhibit binding and activation preference for one or another adrenergic receptor type.
Some agents bind preferably to α-receptors while others exhibit selectivity for β-receptors.
For the case of agonists, binding is associated with receptor activation; therefore, binding selectivity for agonists corresponds to activation selectivity also.
For antagonists the singular event is binding, given that antagonists cannot activate receptors (here the references to "pure" antagonists).
A drug may exhibit receptor selectivity, a non-absolute preference for a particular receptor type.
A drug that is described as "highly specific" will be one that exhibit substantial selectivity for the receptor.
Classification of α-and β -adrenergic receptors with respect to receptor affinities is described below.35
Brief initial comment about some adrenergic agents.
Epinephrine a.k.a. adrenaline activates both α- and β-adrenergic receptors. 35
|
|
The physiological effects actually observed depend in part on dosage. Because the drug activates both adrenergic receptor types, epinephrine at higher doses can cause significant vasoconstriction.
Through cardiac β receptor activation, epinephrine mediates positive inotropic (increased myocardial contractility) and positive chronotropic (increased heart rate) cardiac effects.
Hypertensive responses to epinephrine can be explained both by increased cardiac output as a result of enhanced contractility and heart rate as well as by increased vascular resistance due to α receptor activation.
In a subset of vessels, β2 receptor activation causes vasodilatation.
Skeletal muscle blood vessels respond in this way. Blood pressure effects, then, following epinephrine depend on a balance between α-adrenergic receptor mediated vasoconstriction and β2 adrenergic receptor mediated vasodilatation.
The response to epinephrine may involve both an increase in the systolic pressure (α-adrenergic receptor mediated) along with a decrease in diastolic pressure (reflecting the β2-adrenergic receptor activation). The net effect may be an increase in mean blood pressure with a widening pulse pressure.
Norepinephrine (Levophed) a.k.a. noradrenaline, levarterenol is a principal agonist of both α1 and α2 adrenergic receptors but also activates β1 adrenergic receptors about to the same extent observed with epinephrine.35
|
|
Following norepinephrine infusion, the blood pressure will rise due to prominent α-receptor activation; however, heart rate is likely to decrease, despite norepinephrine activity at β one receptor sites. The reduction in heart rate is explained by an autonomic reflex mediated by baroreceptors (the increased blood pressure causes a parasympathetic (cholinergic)-mediated decrease in heart rate). The cholinergic, parasympathetic response is sufficient to overcome the norepinephrine direct cardiac β1 action.
Some agents are described as direct-acting sympathomimetic drugs-direct activators at the receptors.35
For example, phenylephrine (Neo-Synephrine) is classified as a nearly pure α1 agonist. The absence of the catecholamine structure renders phenylephrine unsuitable as a COMT-substrate (the enzyme catechol-o-methyl transferase thus cannot contribute to phenylephrine inactivation); therefore, phenylephrine exhibits an extended duration of action.
|
|
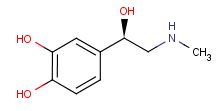
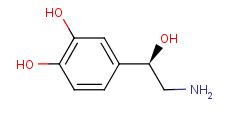
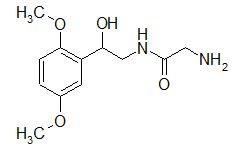
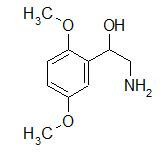
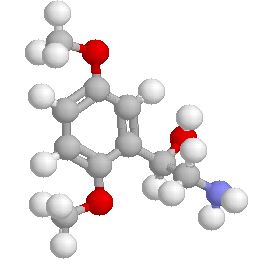
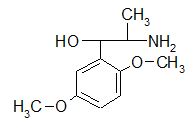
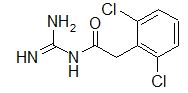
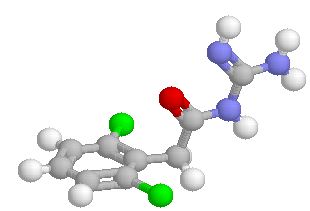
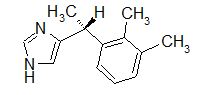
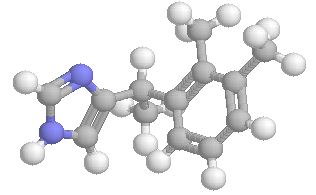
A selective α1 adrenergic agonist, midodrine (Amatine, ProAmatine) is classified as a prodrug.
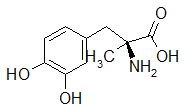
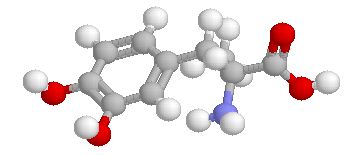
This designation indicates that the physiological response noted after midodrine administration is due to a metabolite, in this case desglymidodrine. Desglymidrine differs from methoxamine only by lacking a CH3 (methyl) group on a side chain. (compared 3D structures below)
Midodrine may be used clinically to manage orthostatic hypotension secondary to and autonomic dysfunction which impairs blood pressure homeostasis.
|
|
|
|
|
|
Methoxamine (Vasoxyl) is another direct acting α1 receptor agonist that both increases blood pressure (α1 mediated vasoconstriction) and bradycardia (reduced heart rate, a parasympathetic-mediated autonomic reflex).
Parasympathetic reflexes that reduce heart rate as described above are mediated by the vagus nerve.35
|
|
|
![]() Other
drugs exhibit relative α2 selectivity and act to
reduce blood pressure as a result of central nervous system
effects35
, perhaps by reducing sympathetic outflow.
Other
drugs exhibit relative α2 selectivity and act to
reduce blood pressure as a result of central nervous system
effects35
, perhaps by reducing sympathetic outflow.
Agents of this type useful in hypertension management include:
Clonidine (Catapres)
Methyldopa (Aldomet)
Guanabenz (Wytensin), and
Guanfacine (Intuniv, Tenex) .
|
|
|
|
|
|
|
|
|
|
|
|
![]() Another
centrally-acting α2-selective agonist which
appears helpful in sedating patients during initial
intubation and subsequent mechanical ventilation is
dexmedetomidine (Precedex).
Another
centrally-acting α2-selective agonist which
appears helpful in sedating patients during initial
intubation and subsequent mechanical ventilation is
dexmedetomidine (Precedex).
|
|
|
Xylometazoline (Otrivin) and oxymetazoline (Afrin), both direct-acting α agonists, are effective topical decongestants which act by constriction of nasal mucosa.35
|
|
|
|
|
|
Isoproterenol (Isuprel) a.k.a. isoprenaline is principally an agonist at β receptors, causing increased myocardial contractility and increased heart rate.35
|
|
|
In addition, its β-effects promote vasodilation sufficient to reduce both mean arterial and diastolic blood pressure, although systolic pressure might be either slightly decreased or slightly increased.35
|
Receptor type: α1 (includes α1A, α1B, α1D) |
example agonist: phenylephrine |
example antagonist: prazosin |
Signal transduction: G protein (Gq,Gi/Go depending on subtype); also depending on subtype, ↑ phospholipase C, D, A2 enzyme activity ↑ IP3, DAG** (true for all α receptor subtypes) |
|
Receptor type: α1 |
Agonist effectiveness: Epinephrine ≥ Norepinephrine >> Isoproterenol; phenylephrine ("pure" α agonist) |
|
|
|
|
*GU--genitourinary; ** IP3: Inositol trisphosphate; DAG: diacylglycerol
IP3:DAG Second Messengers
IP3
DAG
α2 Receptors49,46
Receptor type: α2 (includes α2A, α2B, α2D)
example agonist: clonidine;
α2Aagonist: oxymetazoline
example α2: antagonist: yohimbine;
α2A,α2B: prazosin
Signal transduction: G protein (Gi ↓ adenylyl cyclase activity);
Gi (βγ subunits): ↑K+ channel conductance;
Go : ↓Ca2+ channel conductance (L- and N-type)
↓cAMP (true for all α2 receptor subtypes)
Receptor type: α2
Agonist effectiveness:
Epinephrine ≥ Norepinephrine >> Isoproterenol;
clonidine (classical α2 agonist)
Tissue Effects following α2-receptor activation
Platelets: aggregation
Pancreatic islet β cells: reduction in insulin release
Synaptic endings: reduction in norepinephrine release
Vascular smooth muscle: contraction
β Receptors49,46
Receptor type: β (includes β1, β2, β3)
example agonist: isoproterenol;
β1 agonist: dobutamine;
β2 agonist : albuterol
example β: antagonist: propranol;
β1 antagonist: bextaxolol, metaprolol;
β2 antagonist: butoxamine
Signal transduction: G protein (Gs)
↑ cAMP (true for all β receptor subtypes),
↑adenylyl cyclase,↑ L-type Ca2+ channels
Receptor type: β1
Agonist effectiveness:
Isoproterenol > Epinephrine = Norepinephrine;
dobutamine (antagonist: CGP 20712A)
Tissue Effects following β1 -receptor activation
Renal: juxtaglomerular cells: increased renin secretion
Cardiac: increased myocardial contractility (positive inotropism)
increased rate of contraction
increased AV nodal conduction velocity
Receptor type: β2
Agonist effectiveness:
Isoproterenol > Epinephrine >> Norepinephrine (antagonist ICI 118551);
terbutaline (antagonist: CGP 20712A)
Tissue Effects following β2-receptor activation
Smooth muscle (including bronchial, vascular, GI and GU): relaxation
Skeletal muscle: glycogenolysis and increased K+ uptake.
Hepatic effects:: glycogenolysis and gluconeogenesis
Receptor type: β3
Agonist effectiveness:
Isoproterenol = Epinephrine > Norepinephrine (antagonist ICI 118551);
BRL 37344 (antagonist: CGP 20712A)
Tissue Effects following β3-receptor activation
Adipose: lipolysis
Dopamine Receptors49,46
Receptor type: Dopamine (includes D1, D2,D3, D4,D5 )
example D1 agonist: fenoldopam;
example D2 agonist: bromocriptine
example D4: antagonist: clozapine
Signal transduction:
D1:↑ cAMP;
D2-D5: ↓cAMP
|
|
|
|
|
|
|
|
|
This Web-based pharmacology and disease-based integrated teaching site is based on reference materials, that are believed reliable and consistent with standards accepted at the time of development. Possibility of human error and on-going research and development in medical sciences do not allow assurance that the information contained herein is in every respect accurate or complete. Users should confirm the information contained herein with other sources. This site should only be considered as a teaching aid for undergraduate and graduate biomedical education and is intended only as a teaching site. Information contained here should not be used for patient management and should not be used as a substitute for consultation with practicing medical professionals. Users of this website should check the product information sheet included in the package of any drug they plan to administer to be certain that the information contained in this site is accurate and that changes have not been made in the recommended dose or in the contraindications for administration. Advertisements that appear on this site are not reviewed for content accuracy and it is the responsibility of users of this website to make individual assessments concerning this information. Medical or other information thus obtained should not be used as a substitute for consultation with practicing medical or scientific or other professionals. |