|
|
|
|
|
|
|
Medical Pharmacology Chapter 33-34: Anticancer Drugs
|
|
|
|
|
|
|
Antimetabolites
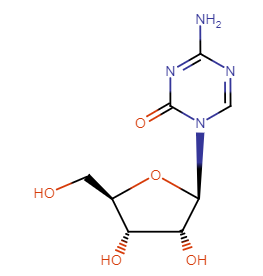
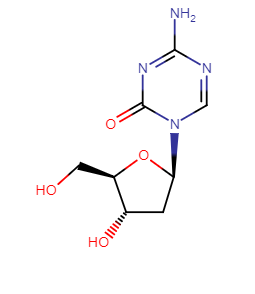
Cytidine Analogues: Azacitidine (Vidaza), decitabine (Dacogen)
 |
 |
 |
 |
5-azacitidine and decitabine (2'-deoxy-5-azacytidine) exert antileukemic activity.1
These agents inhibit DNA cytosine methyltransferase activity and as a result, induce differentiation.
The drugs are approved for myelodysplasia treatment.1
|
 |
|
 |
|
Administration of these agents result in bone marrow normalization in about 15%-20% of patients.
5-azacytidine has been shown to enhanced survival.
Transfusion requirements are reduced in about one-third of patients.1
![]() Myelodysplasia (myelodysplastic syndrome, MDS)7
Myelodysplasia (myelodysplastic syndrome, MDS)7
![]() MDS
represents a variety of hematologic disorders defined by cytopenias
secondary to bone marrow failure as well as by and associated
elevated risk for acute myelogenous leukemia (AML) development.7
MDS
represents a variety of hematologic disorders defined by cytopenias
secondary to bone marrow failure as well as by and associated
elevated risk for acute myelogenous leukemia (AML) development.7
Anemia, with attendant thrombocytopenia withneutropenia frequently, develops with abnormal appearing in usually cellular bone marrow, indicative of ineffective blood cell production.7
Patients described as having "low-risk" myelodysplastic syndrome (MDS) show marrow failure dominating the clinical condition.7
However, in other individuals, myeloblasts are noted at diagnosis along with chromosomal abnormalities.
There is also an associated high-risk for leukemic progression.
![]() Death due to my myelodysplastic syndrome occurs most frequently is a
complication of pancytopenia or leukemia.7
Death due to my myelodysplastic syndrome occurs most frequently is a
complication of pancytopenia or leukemia.7
Many
patients succumb to concurrent diseases, often comorbidities expected
in an elderly population.
Cell Entry:11
Azacytidine enter cells utilizing the ENT1 transporter.
ENT1 is a sodium-independent purine and pyrimidine nucleoside transporter.
Nucleoside transporters are subdivided into two classes:
Equilibrative bi-directional facilitators (ENTs) and
Na+ -dependent concentrative transporters (CNTs).
ENT1 has been described as a glycosylated protein with 456 amino acid residues (50 kDa)
ENTs are proposed to consist of 11-transmembrane helix topology.
ENT1 is a widely distributed transporter, present in erythrocytes, liver, heart, spleen, lung, kidney, intestine and brain.
ENT1 is also expressed and functional in mitochondrial membrane.
Given its tissue distribution, this transporter likely plays an important role in nucleoside provision either derived from diet or produced by tissues, as well as for salvage pathways of nucleotide synthesis and in those cells they do not exhibit de novo synthesis pathways.
ENT1 is thought involved in absorption, distribution and xenobiotic excretion.
![]() Clinically,
ENT1 is important in describing nucleoside analog drug disposition11 .
Clinically,
ENT1 is important in describing nucleoside analog drug disposition11 .
These drugs are part of treatment protocols for cancer, cardiovascular disease, viral and parasitic infections and in certain neurological pathologies.
The drugs in question act following nucleic acid incorporation and typically interfere with nucleic acid synthesis or interfere with physiological nucleoside metabolism.
The first nucleoside analog approved for cancer therapy was the pyrimidine analog cytarabine (ara-C).
This drug is primarily dependent on ENT1 for cell entry.
ENT1 appears responsible for the transport of a number of anticancer drugs into the cell.
These drugs include:
Gemcitabine
5-FU
Pentostatin, and
Cladribine.11
Antineoplastic activity of azacytidine is likely due to drug-induced DNA hypomethylation as well as direct cytotoxic action on abnormal hematopoietic cells localized to the bone marrow. 8
At concentrations required for maximum inhibition of DNA methylation (in vitro), DNA synthesis itself is not substantially suppressed.
Hypomethylation is thought to cause normalization in genes controlling both differentiation and proliferation.8
![]() Following incorporation into DNA, azacytidine inhibits
reversibly DNA methyltransferase.9,10
Following incorporation into DNA, azacytidine inhibits
reversibly DNA methyltransferase.9,10
Inhibition of this enzyme prevents DNA methylation.
It is suggested that an antitumor activity of azacytidine occurs as a result of DNA hypomethylation which activates tumor suppressor genes that had been previously inhibited by hypermethylation.9,10
5-azacytidine is cytotoxic particularly to rapidly dividing cells and exhibit cytotoxicity to malignant cells no longer responding to typical growth control mechanisms.
However, non-proliferating cells are substantially less sensitive to azacytidine.
Following uptake into cells, azacytidine is first phosphorylated to 5-azacytidine monophosphate by uridine-cytidine kinase.
Then, a diphosphate derivative is formed by pyrimidine monophosphate kinase activity.
Finally, the triphosphate derivative is formed utilizing diphosphate kinases to catalyze the reaction.
![]() 5-azacytidine
triphosphate interferes with nuclear and cytoplasmic RNA
metabolism, inhibiting protein synthesis, following its
incorporation into RNA.
5-azacytidine
triphosphate interferes with nuclear and cytoplasmic RNA
metabolism, inhibiting protein synthesis, following its
incorporation into RNA.
![]() Nucleotide
reductase catalyzes a reaction in which 5-azacytidine
diphosphate is converted to 5-aza-deoxycytidine diphosphate.8
Nucleotide
reductase catalyzes a reaction in which 5-azacytidine
diphosphate is converted to 5-aza-deoxycytidine diphosphate.8
The diphosphate form undergoes an additional phosphorylation step catalyzed by nucleoside diphosphate kininases to form 5-azadeoxycitidine triphosphate.
This triphosphate form, following incorporation into DNA, leads to DNA synthesis inhibition.
With respect to the cell cycle, azacytidine exhibits is main toxicity during the S-phase.8
Absorption, Distribution, Biotransformation, Excretion:1
5-azacytidine, following the subcutaneous route of administration, undergoes deamination catalyzed by cytidine deaminase.
The plasma half-life (t1/2) is about 30 minutes.
Following DNA incorporation, aza-nucleoside effects are often long-lasting.1
![]() Side/Adverse Effects:1
Side/Adverse Effects:1
Myelosuppression
G.I. symptoms (mild)
|
|
|
|
|
|
|