|
|
|
|
|
|
|
Medical Pharmacology Chapter 33-34: Anticancer Drugs
|
|
|
|
|
|
|
Hormonal Anticancer Drugs:
![]() Aromatase
Inhibitors (inhibits conversion of androgens into estrogens)
Aromatase
Inhibitors (inhibits conversion of androgens into estrogens)
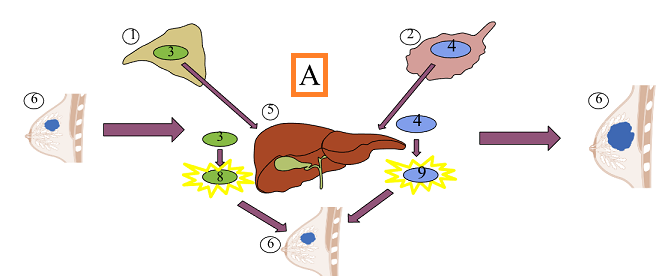
History: The aromatase inhibitors, initially, were administered for management of breast cancer which had metastasized.6
![]() However, at
present (2020), these drugs represent standard treatment for
postmenopausal breast cancer patients.
However, at
present (2020), these drugs represent standard treatment for
postmenopausal breast cancer patients.
Furthermore, aromatase inhibitors may be used as monotherapy both for management of metastatic disease (first-line drug) and as an adjuvant drug.6
The aromatase inhibitors under consideration include anastrozole (Arimidex), letrozole (Femara), and exemestane (Aromasin).13
Anastrozole has shown clinical efficacy in some patients in which breast tumors exhibit resistance to tamoxifen.
Letrozole is comparable in this regard.
These agents are categorized as reversible, selective nonsteroidal aromatase enzyme inhibitors.
Exemestane, steroid, is classified as in irreversible aromatase inhibitor.
Exemestane is similar to anastrozole and letrozole in that it is an agent approved for use in the advanced breast cancer setting.13
The enzyme aromatase catalyzes estrogen formation in several tissue types, including:
Ovarian tissue
Peripheral fat and in
Some tumor cells.
![]() Aromatase (CYP19A1) inhibitors are classified in terms of two types:
Aromatase (CYP19A1) inhibitors are classified in terms of two types:
Type 1: Irreversible steroid analogs (e.g. exemestane) and
Type II: Reversible inhibitors (e.g. anastrazole, letrozole).8
Historically, the rationale for reducing estrogen levels in breast cancer patients presumed functioning ovaries.2
![]() However,
estrogen is also produced by postmenopausal women from
circulating adrenal androgens by aromatization.
However,
estrogen is also produced by postmenopausal women from
circulating adrenal androgens by aromatization.
Noting that estrogen, in premenopausal women, is mainly synthesized in the ovaries:
In premenopausal women aromatase inhibitors induce elevation and gonadotropin synthesis.1
This increase in gonadotropin decreases the ability of aromatase inhibitors to reduce ovarian estrogen synthesis.
Accordingly, aromatase inhibitors would not be effective in premenopausal women unless additional ovarian suppression (for example with GnRH analogs) is provided.1
Sites of nonovarian estrogen synthesis include:
Muscle
Liver as well as
Epithelial and stromal breast tissue.
With advancing age nonovarian estrogens become most prominent.
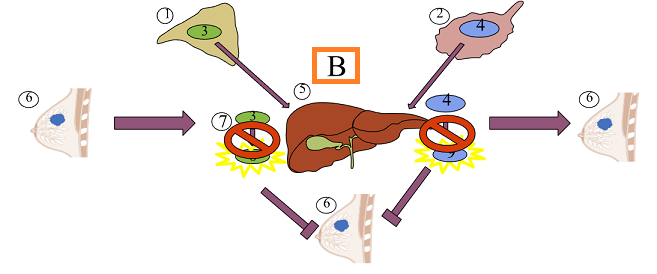
Therefore, since aromatase catalyzes androgen to estrogen conversion, aromatase inhibitors in blocking this process reduce estrogen production.2
![]() Aromatase
inhibitors represent standard of care (adjuvant) in
treating postmenopausal women with estrogen receptor
positive (ER+, presence of estrogen
receptors) breast cancer.1
Aromatase
inhibitors represent standard of care (adjuvant) in
treating postmenopausal women with estrogen receptor
positive (ER+, presence of estrogen
receptors) breast cancer.1
Use of these agents may includes either initial treatment application or administration following tamoxifen use.
Aromatase inhibitors may also be used in initial treatment of metastatic hormone receptor positive (HR+), typically along with a CDK4/6 inhibitor (e.g palbociclib).
Another use of aromatase inhibitors is in the case in which disease progression has occurred in postmenopausal women following tamoxifen treatment.1
|
|
|
|
|
|
|
|
|
|
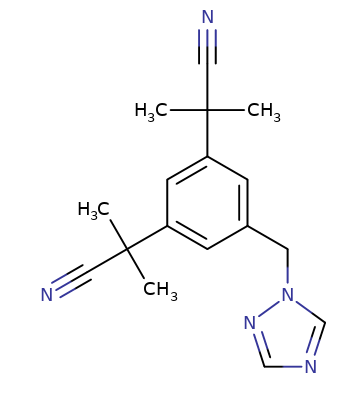
Anastrozole (Arimidex) is a third-generation type II nonsteroidal aromatase (CYP19A1) inhibitor, as is letrozole (Femara).1
The first and second-generation agents such as aminoglutethimide and formestane are not used therapeutically for breast cancer treatment due to side effects.
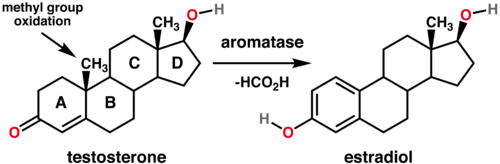
Anastrazole, through aromatase enzyme inhibition, limits the rate of conversion of androstenedione to estrone as well as testosterone to estradiol.17
Changes in aromatase expression may be involved in pathogenesis of estrogen-related cancers (e.g. breast cancer, endometrial cancer).3
However, the third-generation aromatase inhibitors are employed in treating early-stage as well as advanced breast cancer in postmenopausal patients.
|
These agents are also used for chemoprevention.1
Aromatase inhibitors limit estrogen production from androgens by interfering with aromatase enzymic function.9
![]() Limiting
estrone evels by inhibiting conversion of androstenedione to
estrone (aromatase inhibition) as well as testosterone to
estradiol conversion result in: 17
Limiting
estrone evels by inhibiting conversion of androstenedione to
estrone (aromatase inhibition) as well as testosterone to
estradiol conversion result in: 17
Decreasing tumor mass
Delaying tumor progression in those tumors sensitive to hormones.
Anastrazole administration results in a reduction in estrone sulfate concentration by about 85%. 17
Absorption, Distribution, Biotransformation, Excretion:
Following oral administration, anastrozole is easily absorbed by the body.1
Following oral administration, maximum anastrazole drug levels in plasma are reached in about 2 hours.2
In individuals receiving anastrazole at a dosage of 1 mg/d (oral administration) inhibition of aromatase enzymatic activity approaches 97%.
This level of aromatase inhibition substantially reduces estradiol levels (78% to 86%).
Anastrazole half-life ranges from about 38 hours to about 60 hours with steady-state levels reached in about 1 week.
Most pronounced estrogen suppression (maximal) is realized in about 3½ hours following administration.
At the 1 mg/d dosage no effect on gonadotropins or FSH levels have been noted.
Basal or ACTH-stimulated cortisol and aldosterone levels do not appear sensitive to anastrazole administration (even at doses higher than the 1 mg/d level, i.e. 5-10 mg).
Anastrazole binds to plasma protein (40%).2
Anastrozole is metabolized both by phase I (cytochrome P450 mediated drug metabolism) and phase II (conjugation reactions) systems.14
Formation of the hydroxy-anastrozole metabolite, hydroxyanastrozole, is catalyzed mainly by the CYP3A4 cytochrome P450 isoform as well as to a more limited extent by CYP3A5 and CYP2C8 forms.
Subsequently, hydroxyanastrozole is further metabolized through a phase II glucuronidation conjugation pathway.14
A hydroxylated metabolite has also been identified.1
Anastrozole is eliminated mainly by hepatic and biliary systems.
Elimination half-life is estimated to be about two days.1
![]() Anastrazole, through aromatase enzyme inhibition, limits the rate of
conversion of androstenedione to estrone as well as testosterone to
estradiol.2
Anastrazole, through aromatase enzyme inhibition, limits the rate of
conversion of androstenedione to estrone as well as testosterone to
estradiol.2
Anastrazole exerts its therapeutic effect by inhibition of an enzyme, aromatase.3
Aromatase catalyzes conversion of androgens to estrogens.
Aromatase, an enzyme complex, is involved in the final step of estrogen synthesis in which substrates include androgens, androstenedione, and testosterone and products include estrogens and estrone.
Whereas menopause results in cessation of ovarian hormone synthesis, without aromatase inhibition, estrogen may be synthesized by this aromatase catalyzed pathway.3
![]() Anastrazole, an aromatase inhibitor, has been approved for
management of both early-stage breast cancer and for treating
advanced and metastatic disease.1
Anastrazole, an aromatase inhibitor, has been approved for
management of both early-stage breast cancer and for treating
advanced and metastatic disease.1
In early-stage breast cancer anastrazole serves as adjuvant hormonal treatment for an extended period of time, 5-10 years or may be administered after earlier tamoxifen treatment in postmenopausal women (early-stage disease).1
In early-stage breast cancer anastrazole has been found more effective when compared with tamoxifen in extending time before tumor recurrence as well as in reducing the likelihood of primary contralateral tumor occurrence.
In advanced disease, postmenopausal patients exhibiting progressive disease during tamoxifen therapy benefited from anastrozole treatment in terms of survival advantage, comparing anastrazole versus megestrol acetate.
For patients whose metastatic breast cancer disease were estrogen receptor positive (ER+) or progesterone receptor positive (PR+), anastrazole appear to retard disease progression more effectively when compared with tamoxifen.
For those postmenopausal women considered high risk for breast cancer development, anastrazole decreased incidence of invasive disease.3
In premenopausal women less than 35 years old with breast cancer, aromatase inhibitors (e.g. anastrazole) treatment may be combined with ovarian suppression in the adjuvant setting.
Aromatase inhibitors may also be used in women undergoing chemotherapy with aromatase inhibitor administration associated with likely reduced recurrence risk.1
|
Most side effects/adverse effects due to anastrozole administration are related to estrogen depletion.1
![]() Anastrazole
administration in postmenopausal women, is associated with reduced
likelihood many symptoms noted with tamoxifen including:1
Anastrazole
administration in postmenopausal women, is associated with reduced
likelihood many symptoms noted with tamoxifen including:1
Hot flashes
Vaginal discharge
Vaginal bleeding
Endometrial cancer
Ischemic cerebrovascular pathologies
Venous thromboembolic occurrences
Deep venous thrombosis.
Compared to tamoxifen anastrozole administration is more likely be associated with:
Symptomatic arthralgias
Vaginal dryness
Sexual dysfunction.1
![]() Aromatase
inhibitor administration is associated with reduced bone mineral
density.1
Aromatase
inhibitor administration is associated with reduced bone mineral
density.1
Anastrozole administration, compared to tamoxifen, associated with reduced lumbar spine mineral density and reduced total hip mineral density.
Anastrozole administration, compared to tamoxifen, elevates fracture
risk.1
|
|
|
|
Overview: Aromatase inhibitors limit estrogen production from androgens by interfering with aromatase enzymic function.10
|
|
|
Letrozole (Femara) has been demonstrated to be superior compared to tamoxifen in both first-line advanced breast cancer and as a neoadjuvant.6
When compared with anastrazole, Letrozole, in vitro, exhibits greater aromatase enzyme inhibition.
Letrozole, compared to anastrazole, also exhibits greater estrogen suppression both in serum and breast tumor.
In patients with metastatic breast cancer disease, Letrozole appears more effective in second-line endocrine treatment.
Letrozole may not be superior, however, compared to anastrazole, in efficacy or safety in those patients categorized as postmenopausal with hormone receptor-positive, node-positive early-stage breast cancer.6
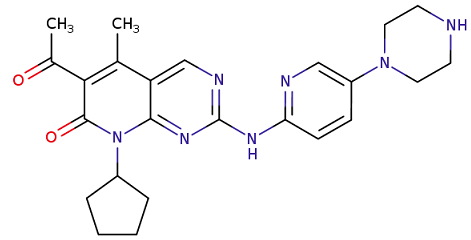
Letrozole may also be administered in combination with a CDK 4/6 inhibitor (Cyclin-Dependent Kinase inhibitor) such as palbociclib (Ibrance) or ribociclib (Kisqali).
Cyclin-dependent kinases (CDKs) 4 and 6 have enzyme activity associated with promoting cell division and proliferation in both normal and malignant cells.
Absorption, Distribution, Biotransformation, Excretion:1,5
Letrozole, following oral administration, exhibits rapid absorption.
The bioavailability is estimated as >99%.
Absorption is not influenced by food.
Steady-state letrozole plasma levels are attained following 2-6 weeks of administration.
Metabolism occurs principally by the cytochrome P450 hepatic microsomal drug metabolism system, involving mainly the CYP2D6 and CYP3A4 isoforms.
The metabolite is mainly an inactive carbinol form.
Half-life of elimination is about two day
Aprroximately 90% of an administered dose is excreted in the urine.
Urinary excretion consists of about 5% unchanged drug, about 9% S unidentified metabolites and about 75% as a glucuronide carbinol metabolite (Phase II metabolism).1,5
Anastrazole, through aromatase enzyme inhibition, limits the rate of conversion of androstenedione to estrone as well as testosterone to estradiol.2
![]() Anastrazole exerts its therapeutic effect by inhibition of an
enzyme, aromatase.3
Anastrazole exerts its therapeutic effect by inhibition of an
enzyme, aromatase.3
Aromatase catalyzes conversion of androgens to estrogens.
|
|
|
Aromatase, an enzyme complex, is involved in the final step of estrogen synthesis in which substrates include androgens, androstenedione, and testosterone and products include estrogens and estrone.
Whereas menopause results in cessation of ovarian hormone synthesis, without aromatase inhibition, estrogen may be synthesized by this aromatase catalyzed pathway.3
In summary, letrozole interaction with the enzyme aromatase results in competitive enzyme inhibition.
Specifically, letrozole interacts with the aromatase theme group.
With respect classification, aromatase is a cytochrome P450 enzyme catalyzing androgen to estrogen conversion (anthracenedione →estrone; testosterone → estradiol) inhibition of aromatase results in reduced plasma estrogen concentration.
Letrozole (Femara) is used in three clinical settings: neoadjuvant, adjuvant and metastatic for treating postmenopausal women with estrogen-positive (ER+) breast cancer.2
Letrozole may be administered in combination with the CDK4/6 inhibitor (palbociclib or ribociclib).
In the clinical trials leading to the approval of these agents (palbociclib and ribociclib) letrozole was chosen to represent the aromatase inhibitor class.
|
|
|
|
![]() As
a result of these clinical trials, palbociclib and ribociclib
were identified as first-line treatment of hormone receptor
positive advanced breast cancer.2
As
a result of these clinical trials, palbociclib and ribociclib
were identified as first-line treatment of hormone receptor
positive advanced breast cancer.2
These clinical use indications for letrozole are those also appropriate for anastrazole administration.
References
|
![]() Adverse
Effects/Toxicities: Similar to those described
above for anastrozole
Adverse
Effects/Toxicities: Similar to those described
above for anastrozole
Exemestane (ex e MES tane, Aromasin)
Aromatase inhibitors limit estrogen production from androgens by interfering with aromatase enzymic function.10
|
|
|
BOLERO-2 Findings Confirm Efficacy of Everolimus Plus Exemestane24
Attribution:
Baselga J BOLERO-2 Findings Confirm Efficacy of Everolimus Plus Exementane https://www.youtube.com/watch?v=tGhgp-oeV64 (1/2012)
Vital Options International Youtube Channel: https://www.youtube.com/channel/UCRSePGODJKDJtjNKk4V4uIA
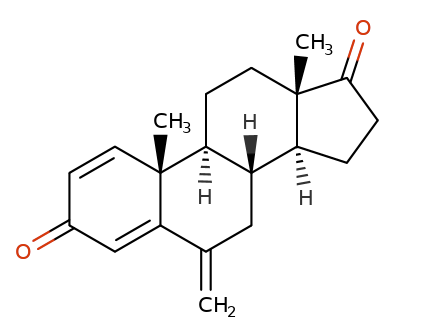
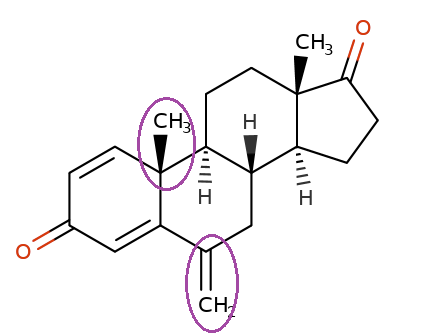
Exemestane (Aromasin) is derived from androstenedione.
Androsteindione
Exemestane
The aromatase active site exhibits very high-affinity for exemestane such that the dissociation of exemestane from the aromatase active site occurs very slowly and the association of exemestane with the aromatase active site occurs very fast.
The very slow dissociation or "off rate" is the basis for exemestane classification as binding "irreversibly" to the enzyme active site.
Because of the very slow dissociation, exemestane effectively inactivates aromatase permanently.2
Exemestane is therefore classified as a steroid-based irreversible (type II) aromatase inhibitor.6
Absorption, Distribution, Biotransformation, Excretion:
Exemestane is orally administered and is rapidly absorbed.1,2
Absorption is about 40% following administration.22
Exemestane is about 90% bound to plasma proteins (mainly serum albumin) and α1-acidic glycoprotein.22
Peak plasma levels are noted about 2 hours following administration.2
High-fat foods tend to increase exemestane absorption and exemestane should probably be administered after eating.
The 40% level of absorption may be increased by about 40% if drug is taken after a high-fat breakfast (compared to fasted state).22
For the 25 mg dose, plasma concentrations fall to below detection levels after about 4 hours.2
However, aromatase inhibition is long-lasting, given that exemestane binds to the enzyme with very high-affinity and associates from the enzyme only very slowly.
Therefore, aromatase enzymic activity is inhibited for at least 5 days following dosing.
Steady-state exemestane levels occur within a week, assuming daily dosing with subsequent estradiol suppression being maximal in the subsequent 3-7 day timeframe.2
The 25 mg exemestane dosage does not significantly affect cortisol, aldosterone, FSH (Follicle-Stimulating Hormone) or LH (Leutinizing-Hormone) levels.2
Exemestane is subject to extensive hepatic metabolism mediated in part by the cytochrome P450 microsomal metabolizing system utilizing primarily the CYP3A4 isoform.2
Exemestane is also metabolized by aldoketoreductases.
Aldoreductase
"Ribbon diagram of human aldose reductase in complex with NADP+, citrate, and IDD594, a small molecule inhibitor."
Attribution: Fvasconcellos (talk · contribs) / Public domain
https://commons.wikimedia.org/wiki/File:Aldose_reductase_1us0.png
Aldoreductases catalyze reduction of aldehydes and carbonyls.2
These enzymes are localized in the cell cytosol in their oxidoreductase activity depends on NADPH.2
Since exemestane is extensively metabolized, many secondary metabolites are formed.
As a consequence of exemestane being excreted in the feces as well as urine, liver and kidney pathology that cause renal and/or hepatic insufficiency may decrease drug clearance and elevate plasma drug levels.2
Also, drug dosage adjustments may be required if exemestane is administered along with a CYP3A4 cytochrome P450 isoform inhibitor, such as rifamicin.6
In this case plasma levels of exemestane were notably reduced.6
Exemestane, steroid-based, acts by inhibiting the enzyme aromatase.6
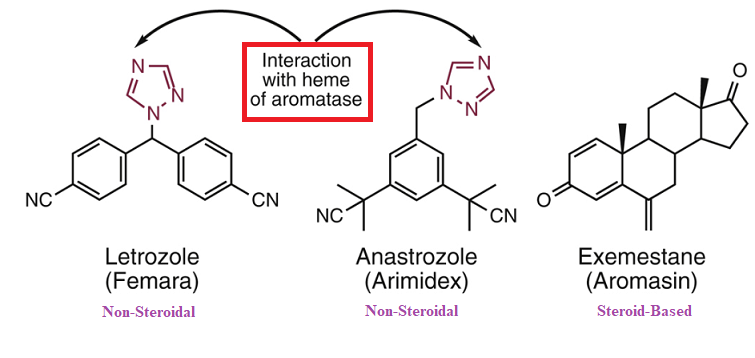
Structural differences between the nonsteroidal and steroidal aromatase inhibitors (letrozole (Femara), anastrazole (Armidex) and exemestane (Aromasin):
Comparison Between Non-Steroidal (Letrozole and Anastrozole) and Steroid-Based (Exemestane) Aromatase Inhibitors6
Attribution:
Adapted from Figure 24.40. Roche VF Cancer and Chemotherapy Chapter 33 in Foye's Principles of Medicinal Chemistry (Roche VG Zito SW Lemke TL Williams DA, eds) Wolters Kluwer | Lippincott Williams & Wilkins, Health, Philadelphia, 8e, 2020. (reference 6).
The inhibition has been classified as irreversible (type II).
Exemestane has been approved for administration as monotherapy in the adjuvant setting following 2-3 years of tamoxifen administration with the aim of completing five consecutive years of adjuvant treatment.6
The setting is hormone receptor positive early breast cancer.6
Exemestane is not approved as first-line adjuvant treatment.
By contrast, letrozole and anastrazole is approved for first-line adjuvant therapy.
Unique to exemestane is its indication for use in combination with everolimus for advanced breast cancer therapy.
Similar to other aromatase inhibitors, everolimus is viewed as appropriate monotherapy in the advanced breast cancer setting, following tamoxifen failure.6
Exemestane is also approved for treating advanced-stage breast cancer which has not been adequately controlled on nonsteroidal type 2 aromatase inhibitors.1
|
|
|
|
|
|
|
.PNG)