Cancer pharmacology begins with a deceptively simple question: why does a drug that kills cancer cells in a dish so often fail to cure cancer in a patient? The answer lies in tumor growth kinetics, the heterogeneity of cell cycle position within a tumor mass, and the relationship between drug exposure, tumor burden, and residual viable cells. Understanding these principles is not academic background material; it is the mechanistic foundation on which every dosing schedule, every combination regimen, and every resistance strategy is built.1
The cell cycle comprises four phases that every dividing cell must traverse: G1 (gap 1, biosynthetic preparation), S (DNA synthesis), G2 (gap 2, premitotic preparation), and M (mitosis). A fifth state, G0, represents a reversible quiescent state outside the active cycle. The duration of G1 is highly variable among cell types and is the primary determinant of overall cycle length; S, G2, and M phases are relatively fixed at approximately 6 to 8 hours, 4 to 6 hours, and 1 hour respectively in most human cancer cell lines. Cells in G0 are metabolically active but not proliferating, and they represent a critical reservoir of drug-resistant cells within a tumor mass because many cycle-specific agents require active DNA synthesis or mitotic spindle assembly to exert their lethal effects.12
The distinction between cycle-specific and cycle-nonspecific agents is central to scheduling decisions. Cycle-specific agents kill only cells that are traversing a particular phase at the time of drug exposure. Antimetabolites such as methotrexate and 5-fluorouracil (5-FU) are S-phase specific; vinca alkaloids are M-phase specific. Because their cytotoxic effect is self-limiting once cells in the sensitive phase have been killed, cycle-specific agents exhibit a plateau on the dose-response curve: beyond the dose that kills all cycling cells in the sensitive phase, further dose escalation does not increase cell kill. This plateau has profound scheduling implications. Cycle-specific agents are best administered as prolonged infusions or repeated dosing to expose cells that enter the sensitive phase after the initial dose. Cycle-nonspecific agents, principally the alkylating agents and the platinum compounds, kill cells regardless of cell cycle position, including G0 cells, and their dose-response relationship is more nearly linear over a wider dose range.12
The log-kill hypothesis, first articulated by Skipper and colleagues in the L1210 (a murine leukemia cell line used as the standard preclinical model) leukemia mouse model, states that a given dose of a chemotherapeutic agent kills a constant fraction, not a constant number, of tumor cells. If a drug kills 99.9% of cells (a 3-log kill) and a patient harbors 10¹⁰ tumor cells at treatment initiation, 10⁷ viable cells remain after the first course. A second equivalent course reduces the burden to 10⁴, and a third to 10¹. This arithmetic predicts that cure requires reducing the tumor burden to less than one viable cell and that early treatment, when tumor burden is lowest, maximizes the probability of cure. The log-kill model also predicts why relapse occurs after apparently complete responses: if the initial tumor burden is 10¹² cells (approximately 1 kg of tumor), even repeated courses achieving 3-log kills will not eradicate the population before resistance emerges or cumulative drug toxicity limits further treatment.2
The Gompertzian growth model refines the log-kill concept by incorporating the observation that real tumors do not grow exponentially throughout their natural history. Early in tumor development, when the mass is small and vascularity is adequate, growth is approximately exponential. As tumor size increases, the growth fraction (the proportion of cells actively cycling) decreases and the doubling time lengthens because nutrient and oxygen supply cannot keep pace with the expanding cell population. At maximum tumor size, the Gompertz curve approaches a plateau. The clinical consequence is that small tumors have the highest growth fraction and are, paradoxically, the most chemosensitive. This is the biological rationale for adjuvant chemotherapy after surgical resection: even when imaging detects no residual disease, microscopic metastatic deposits are growing exponentially with a high growth fraction and are more vulnerable to cell cycle-active drugs than the bulky primary tumor was. The Norton-Simon hypothesis extends this reasoning to scheduling, predicting that dose-dense chemotherapy (the same total dose delivered over a shorter interval) should be more effective than standard scheduling because it prevents tumor regrowth between cycles during the exponential phase of residual disease kinetics.3
For S-phase-specific drugs such as cytarabine (ara-C) in acute myeloid leukemia (AML) induction, the standard regimen uses continuous intravenous infusion over 7 days precisely to expose cells as they enter S phase. A bolus dose given once would kill only the fraction of cells in S phase at that moment (typically 20 to 30% of cycling cells) and miss cells that enter S phase minutes to hours later. Continuous infusion maintains cytotoxic drug concentrations throughout the entire duration of S phase for every dividing cell in the marrow. This scheduling principle, not dose escalation alone, explains why continuous-infusion ara-C produces deeper remissions than equivalent bolus dosing in most AML protocols.
The growth fraction is the proportion of cells within a tumor actively traversing the cell cycle at any given time. Highly aggressive tumors such as Burkitt lymphoma and diffuse large B-cell lymphoma (DLBCL) have growth fractions approaching 90 to 100% and are exquisitely sensitive to cycle-specific agents. Indolent tumors such as follicular lymphoma and chronic lymphocytic leukemia (CLL) have growth fractions below 10% and are comparatively resistant, though they remain responsive to alkylating agents and purine analogs that target cells regardless of cycle position. The growth fraction is not fixed; it changes as tumor burden changes and as cytotoxic therapy kills the cycling population, leaving behind a relatively enriched G0 population. Re-entry of these G0 cells into the cycle during recovery from chemotherapy may paradoxically increase the growth fraction and sensitize residual disease to a subsequent cycle, a phenomenon that has been exploited in some sequential high-dose protocols.13
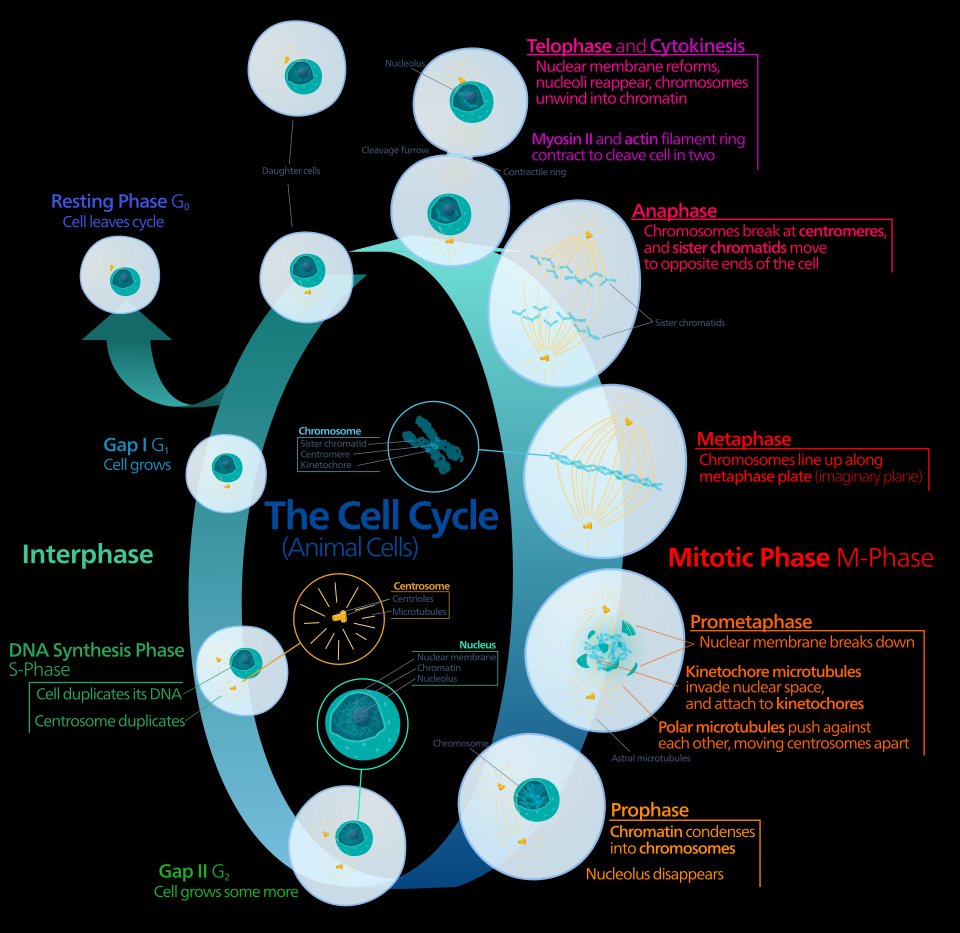
The four active phases of the cell cycle are shown in their sequential relationship: G1 (biosynthetic preparation), S-phase (DNA synthesis and centrosome duplication), G2 (premitotic preparation), and M-phase (mitosis through prophase, prometaphase, metaphase, anaphase, and telophase, completed by cytokinesis). G0 represents the reversible quiescent state. Cells in G0 and those traversing phases other than the drug’s target phase are insensitive to cycle-specific chemotherapy agents, forming the residual viable population that must be addressed by scheduling strategies, combination regimens, or cycle-nonspecific agents.
Kelvinsong, CC0, via Wikimedia Commons.
Oncology pharmacokinetics operates in a clinical environment that amplifies every deviation from textbook drug behavior. Patients present with altered body composition from cachexia, compromised organ function from prior therapy or tumor infiltration, and concurrent medications that rival the chemotherapy regimen in pharmacokinetic complexity. Applying dose adjustment frameworks rigorously is not cautious conservatism; it is the only way to deliver the intended exposure while avoiding preventable, potentially life-threatening toxicity.4
The volume of distribution (Vd) of a cytotoxic drug determines how much drug actually reaches the tumor versus how much is diluted into peripheral tissues. Lipophilic agents such as the nitrosoureas and anthracyclines have very large volumes of distribution, sequestering extensively in adipose tissue and achieving sustained plasma concentrations independent of infusion duration. Hydrophilic agents such as methotrexate and carboplatin have smaller volumes of distribution more closely approximating extracellular fluid volume and are cleared more predictably by renal filtration. In patients with third-space fluid accumulations, such as malignant ascites or large pleural effusions, water-soluble drugs can distribute into these compartments, creating a pharmacokinetic reservoir that slowly releases drug back into the systemic circulation after the plasma concentration has fallen. This delayed release can prolong drug exposure far beyond the expected duration, increasing toxicity without a proportional increase in antitumor effect at the tumor site. Methotrexate is the canonical example: patients with large ascites or effusions require serial drainage before high-dose methotrexate administration to prevent prolonged systemic exposure and mucositis.45
Sanctuary sites are anatomical compartments that are poorly penetrated by systemically administered drugs, creating reservoirs where tumor cells can survive otherwise curative systemic therapy. The central nervous system (CNS) is the most clinically significant sanctuary site. The blood-brain barrier (BBB), formed by tight junctions between cerebral endothelial cells and supported by astrocytic end-feet and pericytes, restricts entry of most hydrophilic and high-molecular-weight drugs. Methotrexate, cytarabine, and thiotepa are among the few conventional agents that achieve therapeutic CNS concentrations after systemic administration; most alkylating agents, anthracyclines, and antimetabolites do not. CNS prophylaxis in high-risk acute lymphoblastic leukemia (ALL) and aggressive non-Hodgkin lymphoma relies on intrathecal drug delivery, direct injection into the cerebrospinal fluid (CSF) via lumbar puncture, to bypass the BBB entirely. The testes represent a second clinically recognized sanctuary site in testicular relapse of ALL in male children, now managed by testicular radiation when relapse occurs. Among the newer small-molecule agents, lipophilicity and molecular weight are the primary structural determinants of CNS penetration, and several tyrosine kinase inhibitors (TKIs) have been specifically engineered for improved BBB penetration to address CNS metastases.5
Protein binding is clinically significant for drugs with narrow therapeutic windows where small changes in free drug concentration translate into large changes in both efficacy and toxicity. Most cytotoxic drugs are moderately to highly protein bound, primarily to albumin. In oncology patients, hypoalbuminemia from malnutrition, hepatic dysfunction, or nephrotic syndrome from paraneoplastic disease is common. A patient with a serum albumin of 2.0 g/dL (normal approximately 4.0 g/dL) may have twice the free fraction of a highly protein-bound drug such as etoposide or paclitaxel, effectively receiving a much higher active drug exposure than the administered dose suggests. Although dose modification based on albumin alone is not standard for most agents, awareness of this phenomenon is essential when toxicity is unexpectedly severe despite a nominally appropriate dose. Conversely, drugs that are not significantly protein-bound, such as carboplatin and methotrexate, are less vulnerable to this source of variability.4
Renal dose adjustment in oncology is most critical for agents excreted predominantly unchanged by glomerular filtration or active tubular secretion. The Calvert formula for carboplatin dosing (dose in mg = target AUC [area under the curve] in mg/mL x min multiplied by [GFR (glomerular filtration rate) + 25]) is the best-established example of pharmacokinetically guided dosing in routine oncology practice. Targeting a specific AUC rather than a weight-based or body surface area-based dose eliminates the systematic underdosing of patients with high GFRs and overdosing of patients with impaired renal function. Methotrexate clearance is almost entirely renal, and any reduction in GFR markedly prolongs plasma half-life, increasing the risk of mucositis and myelosuppression. Concomitant use of nonsteroidal anti-inflammatory drugs (NSAIDs) or proton pump inhibitors (PPIs) with methotrexate reduces renal tubular secretion, and this interaction is dose-dependent and potentially severe at high-dose methotrexate regimens. Cisplatin nephrotoxicity compounds the problem by reducing GFR itself, necessitating dose reduction or substitution of carboplatin in subsequent cycles when cisplatin-related nephrotoxicity develops.45
Hepatic dose adjustment is required for drugs cleared primarily by hepatic metabolism and biliary excretion. The anthracyclines, vinca alkaloids, taxanes, and irinotecan all require dose reduction in the setting of hepatic impairment, typically guided by serum bilirubin as a surrogate for biliary excretory function. The Child-Pugh score, developed for chronic liver disease, provides a composite assessment of hepatic synthetic and excretory function and is frequently used in oncology to guide dose modification, though it was not specifically validated for chemotherapy dosing. For agents such as doxorubicin, guidelines generally recommend a 50% dose reduction when total bilirubin is 1.2 to 3.0 mg/dL and a 75% reduction when bilirubin exceeds 3.0 mg/dL. These thresholds reflect practical risk management rather than precisely derived pharmacokinetic targets, and the oncologist must balance the risk of undertreating the malignancy against the risk of unpredictable severe toxicity from impaired drug clearance.4
Four scenarios disproportionately account for serious chemotherapy toxicity attributable to pharmacokinetic derangements: (1) administering NSAIDs or PPIs concurrently with high-dose methotrexate, delaying clearance and causing life-threatening mucositis; (2) giving methotrexate to a patient with unrecognized large pleural effusion or ascites without prior drainage; (3) using weight-based carboplatin dosing in a patient with impaired renal function instead of the Calvert AUC-based formula, resulting in a twofold or greater overdose; and (4) administering full-dose anthracyclines or vinca alkaloids to a patient with significant hyperbilirubinemia. Each of these errors is preventable with pre-treatment pharmacokinetic assessment.
Drug resistance is the central unsolved problem of clinical oncology. Most patients who relapse after initial chemotherapy do so with disease that is resistant not only to the agents used in initial treatment but also to structurally unrelated drugs, a phenomenon called multidrug resistance (MDR). Understanding the specific molecular mechanisms of resistance is not merely of academic interest; each mechanism predicts which rescue strategies are likely to succeed, which combinations may prevent resistance from emerging, and which molecular tests can be used to identify resistant disease prospectively.6
The most extensively characterized resistance mechanism is overexpression of P-glycoprotein (P-gp), the protein product of the MDR1 (multidrug resistance 1) gene (also designated ABCB1 [ATP-binding cassette subfamily B member 1], a member of the ATP [adenosine triphosphate]-binding cassette [ABC] transporter superfamily). P-gp is an energy-dependent efflux pump embedded in the plasma membrane that uses ATP hydrolysis to actively transport a broad range of structurally diverse hydrophobic compounds out of the cell, reducing intracellular drug concentrations below cytotoxic thresholds. Its natural substrate range includes bile acids, steroid hormones, and xenobiotics, but it avidly transports anthracyclines, vinca alkaloids, taxanes, epipodophyllotoxins (etoposide, teniposide), and many tyrosine kinase inhibitors. P-gp is constitutively expressed at high levels in many normal tissues including the intestinal epithelium, the blood-brain barrier (BBB), hepatocytes, and renal tubular cells, where it serves a protective barrier function. Tumor cells can acquire P-gp overexpression through gene amplification, promoter hypomethylation, or selection of pre-existing P-gp-high clones during chemotherapy exposure. Numerous P-gp inhibitors have been evaluated in clinical trials, but none has improved outcomes without unacceptable pharmacokinetic interactions, partly because P-gp inhibition also blocks drug efflux from normal tissues and increases systemic drug exposure requiring dose reduction.67
Beyond P-gp, additional ABC family transporters contribute to MDR. MRP1 (ABCC1), the multidrug resistance-associated protein 1 (encoded by the ABCC1 gene), transports organic anions and glutathione conjugates and is particularly relevant to resistance to anthracyclines, vinca alkaloids, and methotrexate. BCRP (ABCG2), the breast cancer resistance protein, is highly expressed in hematopoietic stem cells and confers resistance to mitoxantrone, camptothecin derivatives (irinotecan, topotecan), and several oral kinase inhibitors including imatinib and gefitinib. The simultaneous overexpression of multiple ABC transporters in relapsed and refractory disease creates a broad-spectrum drug extrusion system that current pharmacological inhibition strategies cannot overcome without prohibitive toxicity.6
Altered drug metabolism constitutes a second category of resistance mechanisms. For prodrugs that require intracellular activation, reduced expression of the activating enzyme confers resistance. Cytarabine resistance in AML (acute myeloid leukemia) frequently involves downregulation of deoxycytidine kinase, the enzyme responsible for phosphorylating cytarabine to its active triphosphate form (ara-CTP). 6-mercaptopurine (6-MP) resistance involves loss of hypoxanthine-guanine phosphoribosyltransferase (HGPRT), the enzyme that converts 6-MP to its active nucleotide form. Conversely, for drugs that must remain in their administered form to be active, upregulation of catabolic enzymes confers resistance. Dihydropyrimidine dehydrogenase (DPD), the rate-limiting enzyme in 5-FU catabolism, is overexpressed in some colorectal tumors and correlates with clinical 5-FU resistance. The same enzyme, when genetically deficient (DPD deficiency affecting approximately 3 to 8% of the population), is responsible for severe and potentially fatal 5-FU toxicity in patients who receive standard doses, illustrating that the same metabolic enzyme can be a resistance mechanism at one extreme and a toxicity determinant at the other.7
Target amplification or mutation directly reduces drug binding to its molecular target. Dihydrofolate reductase (DHFR) gene amplification is the prototypic mechanism of methotrexate resistance: the tumor cell produces so much DHFR enzyme that therapeutic methotrexate concentrations cannot inhibit all of it, and the residual uninhibited fraction is sufficient to maintain folate metabolism and DNA (deoxyribonucleic acid) synthesis. Topoisomerase II mutations alter the drug-binding domain of the enzyme, reducing etoposide and anthracycline binding without eliminating essential enzymatic function. Among the platinum compounds, reduced expression of copper transporter 1 (CTR1), which mediates cisplatin uptake, combined with increased expression of copper-exporting transporters ATP7A (ATPase copper transporting alpha) and ATP7B (copper-transporting ATPases that pump cisplatin out of the cell) reduces intracellular cisplatin accumulation. Downstream target mutation is most dramatically illustrated by BCR-ABL (breakpoint cluster region-Abelson kinase) kinase domain mutations in imatinib-resistant chronic myeloid leukemia (CML), which represent a paradigm for targeted therapy resistance and are discussed in detail in Chapter 34.67
Defects in apoptosis signaling pathways represent a final and particularly important category of resistance. Most cytotoxic drugs kill cells by inducing apoptosis, the programmed cell death cascade. Loss of functional p53 is present in approximately 50% of human cancers and eliminates the primary transcription factor responsible for upregulating pro-apoptotic genes (BAX, PUMA, NOXA) in response to DNA damage. Without p53-mediated apoptotic signaling, cells with drug-induced DNA damage arrest but do not die, allowing time for DNA repair and eventual resumption of proliferation. Overexpression of BCL-2 (B-cell lymphoma 2 protein, the founding anti-apoptotic family member), and its relatives BCL-XL (BCL-2-extra large) and MCL-1 (myeloid cell leukemia 1), tilts the balance at the mitochondrial outer membrane toward cell survival by sequestering pro-apoptotic proteins BIM (BCL-2-interacting mediator of cell death), BAX, and BAK (BCL-2 antagonist/killer). BCL-2 overexpression was first identified in follicular lymphoma through the t(14;18) chromosomal translocation that places the BCL-2 gene under immunoglobulin heavy chain promoter control; it subsequently became a therapeutic target addressed by venetoclax, a BCL-2-selective BH3 (BCL-2 homology domain 3) mimetic that is discussed in Chapter 34.613
Given that resistance mechanisms are varied, often pre-exist in tumor subclones before treatment begins, and emerge rapidly under drug selection pressure, the strategy of combining agents with non-overlapping mechanisms of action reduces the probability that any single resistant subclone will possess resistance to all drugs in the regimen simultaneously. If the frequency of resistance to drug A is 10-6 and to drug B is 10-6, the probability of simultaneous resistance to both is approximately 10-12, below the tumor cell number present even in microscopic disease. This multiplicative probability argument is the mathematical foundation of combination chemotherapy and predicts that regimens with more mechanistically distinct components should theoretically suppress resistance emergence more effectively, within the limits imposed by combined toxicity.
The era of curative chemotherapy began with combination regimens. The individual drugs in MOPP (mechlorethamine, vincristine [Oncovin], procarbazine, prednisone), CHOP (cyclophosphamide, doxorubicin [hydroxydaunorubicin], vincristine [Oncovin], prednisone), and BEP (bleomycin, etoposide, cisplatin [Platinol]) were each inadequate as monotherapy for the diseases they cure; it was their combination, guided by the principles of non-overlapping toxicity and non-cross-resistant mechanisms, that transformed Hodgkin lymphoma, aggressive B-cell lymphoma, and testicular germ cell tumors from uniformly fatal diseases into predominantly curable ones.38
The first principle of rational combination chemotherapy design is that each agent should have demonstrated single-agent activity against the target tumor type. Adding an ineffective drug to a regimen does not improve outcomes and adds toxicity. The second principle is non-overlapping dose-limiting toxicity: the drugs in the combination should have different organs at risk for their most severe adverse effects, allowing each to be given at its full single-agent dose or close to it. CHOP exemplifies this principle. Cyclophosphamide dose-limits on myelosuppression; doxorubicin dose-limits on myelosuppression and cumulative cardiotoxicity; vincristine dose-limits on peripheral neurotoxicity with minimal myelosuppression; prednisone contributes anti-lymphoma activity through glucocorticoid receptor-mediated apoptosis in lymphoid cells and anti-inflammatory support with essentially no myelosuppression. The four drugs together deliver four independent mechanisms of action (alkylation, intercalation plus topoisomerase II inhibition, tubulin depolymerization, glucocorticoid receptor activation) with partially non-overlapping toxicity profiles, achieving a combined effect far superior to any single agent alone.3
MOPP was the first regimen to cure Hodgkin lymphoma, establishing proof of concept for combination chemotherapy in 1970. It combined mechlorethamine and procarbazine as alkylating agents (different chemical classes, reducing cross-resistance), vincristine for mitotic arrest (minimal myelosuppression at standard doses), and prednisone for lymphocytotoxic and anti-inflammatory effects. MOPP achieved complete remissions in approximately 80% of advanced Hodgkin lymphoma patients and long-term cures in about 50%, an unprecedented result at the time. It was subsequently supplanted by ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) because ABVD produced equivalent or superior cure rates with substantially lower risks of secondary leukemia and infertility, both serious long-term MOPP toxicities attributable to its alkylating agent components. The MOPP-to-ABVD transition remains a textbook example of how refinement of combination regimens based on long-term toxicity data can improve the therapeutic index of curative therapy without sacrificing efficacy.8
BEP for testicular germ cell tumors illustrates the principle that even metastatic solid tumors can be curable with combination chemotherapy if the biology is favorable. Cisplatin provides the backbone alkylating-like activity through platinum-DNA adduct formation; etoposide adds topoisomerase II inhibition; bleomycin contributes DNA (deoxyribonucleic acid) strand-break induction through iron-mediated free radical generation. The combination achieves cure rates exceeding 80% even in patients with disseminated disease, a result attributable to the inherent chemosensitivity of germ cell tumor biology rather than to any single agent. The regimen's limitations are defined by the severe acute toxicity of high-dose cisplatin (nephrotoxicity, ototoxicity, nausea) and the cumulative pulmonary toxicity of bleomycin, which mandates the strict bleomycin cumulative dose limit and the avoidance of supplemental high-concentration oxygen in the perioperative period for any patient who has received bleomycin.8
Dose intensity, defined as the amount of drug delivered per unit time (usually expressed in mg/m²/week), is a critical determinant of outcome in responsive tumors. Retrospective analyses of breast cancer adjuvant trials demonstrated that patients who received less than 85% of their planned dose intensity had significantly worse disease-free and overall survival than those who received full-dose therapy. This finding motivated the development of granulocyte colony-stimulating factor (G-CSF) support to allow full-dose delivery by accelerating neutrophil recovery after myelosuppressive chemotherapy. The practical implication is that arbitrary dose reductions for hematologic toxicity, when G-CSF support could instead allow full-dose delivery, may compromise the curative potential of adjuvant or potentially curative regimens and should be avoided when clinically feasible.39
Dose-dense chemotherapy applies the Norton-Simon hypothesis by compressing the interval between cycles from three weeks to two weeks, maintaining the same per-cycle dose but increasing dose intensity by reducing the recovery time during which residual tumor cells can proliferate. The landmark CALGB (Cancer and Leukemia Group B) 9741 trial demonstrated that dose-dense AC-T (doxorubicin and cyclophosphamide followed by paclitaxel) with G-CSF support improved disease-free and overall survival compared with the same regimen given on a standard three-week schedule in node-positive breast cancer. The biological rationale is that compressing the inter-cycle interval prevents exponential regrowth of residual microscopic disease during the recovery period when the growth fraction of surviving cells is highest. Dose-dense scheduling requires G-CSF support to be practical and safe; without it, the shortened interval would produce cumulative myelosuppression that would force delay or dose reduction, negating the intended intensification.9
Dose intensity refers to the rate of drug delivery (mg/m²/week) and can be increased either by raising the per-cycle dose or by shortening the interval between cycles. Dose density specifically refers to shortening the inter-cycle interval while maintaining the per-cycle dose. High-dose chemotherapy with stem cell rescue increases dose intensity through massive per-cycle dose escalation, sometimes tenfold above standard doses, relying on autologous or allogeneic stem cell infusion to rescue marrow function. Dose-dense chemotherapy achieves a more modest intensity increase (approximately 33% by compressing three-week to two-week cycles) that is achievable with G-CSF support alone. The two strategies are not interchangeable and apply to different clinical settings with different supporting evidence.
Chemotherapy drugs impose risks not only on patients but also on the healthcare workers who prepare and administer them. Safe handling standards and extravasation management protocols are not procedural formalities; they represent evidence-based frameworks for preventing irreversible tissue injury in patients and occupational genotoxic exposure in clinicians, nurses, and pharmacy staff.10
Intravenous chemotherapy agents are classified by their potential to cause tissue damage if they leak from the vein into surrounding tissue during administration. Vesicants cause severe tissue destruction, blistering, and potentially deep necrosis that may require surgical debridement or skin grafting. The most important clinical vesicants are the anthracyclines (doxorubicin, daunorubicin, epirubicin, idarubicin), the vinca alkaloids (vincristine, vinblastine, vinorelbine), mechlorethamine, mitomycin C, and paclitaxel. Among these, the anthracyclines are particularly dangerous because doxorubicin binds to tissue DNA (deoxyribonucleic acid) and is released slowly from necrotic tissue, causing progressive local injury that continues to advance for weeks after the initial extravasation event. Irritants cause pain, inflammation, and phlebitis at and proximal to the injection site but do not cause tissue necrosis. Irritants include carboplatin, cisplatin, etoposide, ifosfamide, and 5-FU at standard concentrations. The distinction between vesicant and irritant is not always absolute; concentrated solutions of nominally irritant drugs can behave as vesicants, and patient tissue factors such as poor subcutaneous tissue from prior surgeries or radiation can amplify injury from irritants.10
The management of vesicant extravasation depends on the offending agent. General measures for all extravasations include immediately stopping the infusion, aspirating residual drug through the existing catheter before removal, and marking the affected area for monitoring. For anthracycline extravasation, dexrazoxane (Totect) is the only FDA-approved antidote and should be administered intravenously within six hours of the extravasation event. Dexrazoxane chelates iron, reducing the iron-mediated free radical generation that drives anthracycline tissue toxicity, and also inhibits topoisomerase II-mediated DNA damage in the affected tissue. In the absence of dexrazoxane, dimethyl sulfoxide (DMSO) topical application has been used as an alternative. For vinca alkaloid extravasation, warm compresses and hyaluronidase local injection are recommended; unlike anthracyclines, cooling is contraindicated for vincas because it promotes crystallization in local tissue. For mechlorethamine extravasation, sodium thiosulfate injection neutralizes the alkylating agent directly in tissue. All vesicant extravasations should be documented formally, photographed, and followed with surgical consultation if tissue necrosis develops.10
The NIOSH (National Institute for Occupational Safety and Health) hazardous drug list classifies drugs meeting criteria for carcinogenicity, genotoxicity, reproductive toxicity, teratogenicity, developmental toxicity, or organ toxicity at low doses. Essentially all cytotoxic chemotherapy agents qualify. NIOSH standards mandate closed-system drug transfer devices (CSTDs) for hazardous drug preparation, biological safety cabinet or isolator preparation by pharmacy, double gloving and gown use for all handling, and respiratory protection during any procedure that could generate aerosols. Healthcare workers who are pregnant, attempting conception, or breastfeeding should not handle hazardous drugs. The oncology community has been slow to implement comprehensive NIOSH compliance in all practice settings, and environmental monitoring studies continue to detect hazardous drug surface contamination in chemotherapy preparation areas, patient rooms, and even home environments of patients undergoing oral chemotherapy.14
Anthracycline extravasation is a genuine oncologic emergency. The six-hour window for dexrazoxane administration is not flexible; delays beyond six hours substantially reduce its efficacy in preventing tissue necrosis. If dexrazoxane is not immediately available on the unit, pharmacy must be contacted simultaneously with cessation of the infusion. Do not apply cooling; topical cooling is contraindicated with dexrazoxane (it reduces local blood flow and dexrazoxane delivery) and is not the preferred management for anthracyclines even in settings where dexrazoxane is unavailable. Document the volume and concentration of drug extravasated, the time of discovery, and all subsequent interventions, as this information guides surgical planning if necrosis develops.
The drugs used to prevent and manage the complications of chemotherapy are themselves a sophisticated pharmacological system. Supportive care pharmacology determines whether a patient can complete planned chemotherapy at full dose intensity, whether febrile neutropenia is managed safely, and whether tumor lysis syndrome is recognized and treated before it produces life-threatening metabolic consequences. Mastery of supportive care agents is as essential to oncology practice as mastery of the cytotoxic drugs themselves.11
Febrile neutropenia (FN) is defined as a temperature above 38.3 degrees Celsius (101 degrees Fahrenheit) as a single measurement or above 38.0 degrees Celsius (100.4 degrees Fahrenheit) sustained over one hour in a patient with an absolute neutrophil count (ANC) below 500 cells/mcL or below 1,000 cells/mcL and expected to fall below 500 cells/mcL within 48 hours. It carries a mortality risk of 5 to 10% in unselected oncology patients and much higher rates in patients with ANC below 100 cells/mcL, prolonged neutropenia duration exceeding 10 days, or significant comorbidities. Management requires prompt broad-spectrum antibacterial coverage initiated within one hour of presentation. For low-risk FN (solid tumors, ANC expected to recover within 7 days, no significant comorbidities, MASCC [Multinational Association for Supportive Care in Cancer] score above 21), oral fluoroquinolone plus amoxicillin-clavulanate outpatient management is appropriate. High-risk FN requires hospital admission and intravenous anti-pseudomonal beta-lactam therapy, most commonly piperacillin-tazobactam as first-line in most institutional protocols, with carbapenem reserved for patients with prior resistant organisms or clinical deterioration.1115
Granulocyte colony-stimulating factors (G-CSFs) are recombinant cytokines that stimulate neutrophil precursor proliferation and differentiation in the bone marrow, shortening the duration of chemotherapy-induced neutropenia by three to five days and reducing the incidence of FN by approximately 50% in high-risk regimens. Filgrastim is a non-glycosylated recombinant human G-CSF (rhG-CSF) with a short half-life of approximately 3.5 hours requiring daily subcutaneous injection beginning 24 to 72 hours after the last dose of chemotherapy. Pegfilgrastim is filgrastim conjugated to a 20-kilodalton polyethylene glycol molecule; the PEGylation dramatically reduces renal clearance (the primary elimination route of filgrastim) and prolongs half-life to approximately 33 hours, allowing single-injection per-cycle dosing. Lipegfilgrastim is a more recently approved glycoPEGylated G-CSF with similar pharmacokinetics to pegfilgrastim. G-CSF should not be started within 24 hours of chemotherapy because it mobilizes proliferating neutrophil precursors that become vulnerable to cycle-specific cytotoxic agents if drug levels are still elevated, potentially worsening myelosuppression. The primary adverse effects of G-CSF are bone pain from marrow expansion (managed with antihistamines, NSAIDs, or low-dose opioids) and, rarely, splenic enlargement and rupture, a serious complication that has been reported primarily in healthy donors undergoing G-CSF mobilization for stem cell donation.911
Chemotherapy-induced nausea and vomiting (CINV) is classified by its temporal relationship to chemotherapy administration and its emetogenic potential. Acute CINV occurs within 24 hours of chemotherapy and is mediated primarily by serotonin (5-HT3) release from enterochromaffin cells in the gastrointestinal tract, stimulating vagal afferent 5-HT3 receptors and activating the vomiting center. Delayed CINV occurs 24 to 120 hours after chemotherapy and is mediated predominantly by substance P binding to neurokinin-1 (NK-1) receptors in the brainstem, particularly in the nucleus tractus solitarius. Anticipatory CINV is a conditioned response in patients who have experienced prior inadequately controlled CINV and is resistant to antiemetic pharmacotherapy; it is best prevented by achieving optimal CINV control from the first cycle. The antiemetic regimen is selected based on the emetogenic potential of the chemotherapy: highly emetogenic chemotherapy (HEC), including cisplatin, carmustine, cyclophosphamide at doses above 1,500 mg/m², and dacarbazine, requires triple-drug antiemetic prophylaxis with a 5-HT3 receptor antagonist (ondansetron, granisetron, or palonosetron), an NK-1 receptor antagonist (aprepitant or fosaprepitant, or netupitant in the fixed combination NEPA with palonosetron), and dexamethasone. Moderately emetogenic chemotherapy (MEC) such as carboplatin, doxorubicin, and irinotecan requires at minimum a 5-HT3 antagonist plus dexamethasone, with NK-1 antagonist added in guidelines that recognize carboplatin as highly emetogenic in combination regimens.16
Tumor lysis syndrome (TLS) results from the massive and rapid release of intracellular contents into the systemic circulation when large numbers of tumor cells die simultaneously after initiation of cytotoxic therapy. The metabolic consequences are predictable from first principles: hyperuricemia from nucleic acid catabolism (purines are catabolized to hypoxanthine, then xanthine, then uric acid by xanthine oxidase), hyperkalemia from intracellular potassium release, hyperphosphatemia from intracellular phosphate release, and secondary hypocalcemia from calcium-phosphate precipitation. The clinical consequences of established TLS include acute uric acid nephropathy (uric acid crystallization in renal tubules, producing acute kidney injury), life-threatening cardiac arrhythmias from hyperkalemia, and hypocalcemic tetany or seizures. The highest-risk tumors are those with large cell mass and high proliferative rate that are highly chemosensitive: Burkitt lymphoma, acute lymphoblastic leukemia (ALL) with high white blood cell counts, and acute myeloid leukemia (AML) with blast counts above 100,000 cells/mcL.1112
TLS prophylaxis begins before chemotherapy with aggressive intravenous hydration (typically at two to three times maintenance to maximize urinary uric acid excretion and dilute crystallizing anions), allopurinol (a xanthine oxidase inhibitor that blocks new uric acid production, initiated 24 to 48 hours before chemotherapy), and correction of pre-existing electrolyte abnormalities. For patients at high TLS risk (large tumor burden, high LDH [lactate dehydrogenase], baseline hyperuricemia), rasburicase (recombinant urate oxidase) is preferred over allopurinol because it rapidly degrades existing uric acid to allantoin, a highly soluble product that is readily excreted; allopurinol only prevents new uric acid formation and does not lower the pre-existing uric acid burden. Rasburicase is absolutely contraindicated in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency because the hydrogen peroxide generated by the rasburicase reaction causes severe hemolytic anemia in G6PD-deficient erythrocytes.1112
Rasburicase is rapidly becoming standard care for high-risk TLS prophylaxis, but it cannot be given safely without knowing the patient's G6PD status. G6PD deficiency affects approximately 10% of African-American males and 5% of Mediterranean-ancestry males. In G6PD-deficient patients, the hydrogen peroxide generated by the rasburicase-catalyzed oxidation of uric acid to allantoin cannot be neutralized by the glutathione pathway, causing acute intravascular hemolysis that can be severe and life-threatening. G6PD screening should be performed before rasburicase administration in any patient with ancestry from regions where G6PD deficiency is prevalent. In urgent situations where G6PD testing is not yet available, allopurinol plus aggressive hydration is the safer default while results are pending.