Volume of distribution (Vd)
is the ratio between the amount of drug in body
(dose given) and the concentration of the drug
(C) measured in blood or
plasma.
Vd =
(amount of drug in body)/C where C is the concentration of drug
in blood or plasma.
Vd as calculated is an
apparent volume of distribution. For example:
Vd
for digoxin is 440 L/70 kg (liters per 70
kg person)
Vd
for chloroquine is 13,000 L/70 kg (liters
per 70 kg person)
Such very large Vd
would be consistent with very high tissue
binding, leaving little free in plasma or
blood
Vd is an apparent
volume of distribution, since Vd is
the volume needed to contain the amount of drug homogeneously
at the concentration found in the blood, plasma,
or plasma water.
Many drugs have a
much higher concentration in
extravascular compartments (therefore
these drugs are NOT homogeneously
distributed)
Physical volumes
(L./kg body weight) for some body compartments
Water
Total Body Water (0.5-0.7 L/kg)
or about 35000 to 49000 ml (70 kg individual)
Extracellular Water (0.2 L./kg)
Blood (0.08 L./kg);
plasma (0.04 L./kg)
Bone
0.07
L/kg
Semilogarithmic plot above
illustrates extrapolation to time 0 required to determine the volume
of distribution;Vd = dose/Co-
also note that the drug elimination halftime can be directly
calculated from the graph. This graph applied for a single
compartment model only. For multiple compartments which will
appear as a. non-linear relationship extrapolation back to t = 0
must be performed for each compartment separately. From Goodman Gilman, A, Rall T,
Nies, A, Taylor P, eds Goodman and Gillman: The
Pharmacological Basis of Therapeutics, 8th edn, Oxford: Pergamon,
1990
Factors influencing the volume of
distribution:
drug pKa
extent of drug-plasma
protein binding
partition coefficient of
the drug in fat (lipid solubility)
Vd
may be affected by:
patient's
gender
patient's
age
patient's
disease
patient's
body composition
Example of
a poorly lipid soluble agent with a Vd
about equal to extracellular fluid
volume: nondepolarizing neuromuscular
blocking drugs.
Clearance
is especially important for insuring
appropriate long-term drug dosing, important to the
obtaining of correct steady-state drug concentrations
Clearance of
a given drug is usually constant over the
therapeutic concentration range because:
Drug elimination systems
are not saturated -- therefore the
absolute rate of elimination is a linear
function of the drug's plasma
concentration.
Drug elimination is
therefore usually a first-order kinetic
process-- a constant fraction of the drug
is eliminated per unit time.
Some drugs (e.g.,
ethanol) exhibit zero order kinetics --
a constant amount of drug is eliminated
per unit time. {Clearance is variable}
Clearance: the drug's rate of elimination (by all routes)
normalized to the concentration of drug C in some
biological fluid:
CL
= Rate of elimination /
C
CL
= Vd x kel where Vd
= volume of distribution and kel is the elimination rate
constant
CL
= Vd x (0.693/t1/2) where
0.693 = ln2 and t1/2
is the drug elimination half-life
Clearance:
volume per unit time
(volume of fluid i.e. blood or plasma
that would be completely freed of drug to
account for the elimination)
may be defined as:
blood clearance,
CLb
plasma clearance,
CLp
concentration of
unbound or free drug, depending
on the concentration measured (Cb,
Cp or Cu)
Clearance is additive: a function of elimination by all
participating organs such as liver or kidney:
CL systemic = CLrenal
+ CLhepatic + CLother
"Other"
sites may include the lungs and
other sites of drug metabolism
(muscle, blood)
The two most
important sites for drug
elimination: kidneys and liver
Kidneys: most important organs
for unchanged drug/drug
metabolites elimination
Water-soluble compounds exhibit
more
efficient renal excretion
compared to lipid soluble
compounds (emphasizing
the importance of
metabolic conversion of
lipid-soluble drugs to
water-soluble
metabolites)
Renal drug clearance is correlated with
exogenous creatinine clearance or
serum creatinine concentration
Factors in renal
excretion:
Glomerular
filtration-- important
considerations:
Fraction
of free drug (compared to
protein-bound drug)--when
a drug is bound to
protein it is not
filtered
Glomerular
filtration rate
Tubular
secretion (active
process)-- important
considerations:
Drug/metabolite
selectivity
Passive
tubular reabsorption-- important
considerations:
Drug delivery to hepatic
elimination sites may be rate-limiting
for certain drugs:
also
called flow
dependent elimination: in this case most of
the drug in the blood is
eliminated on the first pass of
the drug through the organ
these
drugs are termed
"high-extraction"
extent of plasma
protein-bound drug
blood flow (affects clearance
on drugs with high extraction ratios).
Clearances > 6
ml/min./kg -- including:
chlorpromazine:
(antipsychotic)
diltiazem: (Ca2+
channel blocker)
imipramine:
(tricyclic
antidepressant)
lidocaine:
(antiarrhythmic)
morphine: (opioid
analgesic)
propoxyphene:
(opioid
analgesic)
propranolol: (beta adrenergic receptor
blocker)
verapamil: (Ca2+
channel blocker)
meperidine:
(opioid analgesic)
desipramine:
(tricyclic
antidepressant)
amitriptyline:
(tricyclic
antidepressant)
isoniazid:
(anti-tuberculosis)
Changes
in the intrinsic clearance (i.e. enzyme
induction, hepatic disease: affects clearance
of drugs with low extraction ratios): Examples --
Social factors:
Tobacco smoke induces
some hepatic microsomal drug metabolizing enzyme
isoforms (CYP1A1, CYP1A2, and possibly CYP2E1)
Chronic ethanol use
induces CYP2E1
Dietary
considerations:
Grapefruit juice
contains chemicals that are potent inhibitors of
CYP3A4 localized in the intestinal wall mucosa
Cruciferous
vegetables such as brussels sprouts, cabbage,
cauliflower and hydrocarbons present in
charcoal-broiled meats can induce CYP1A2.
Calcium present in
dairy products can chelate drugs including commonly
used tetracyclines and fluoroquinone
antibiotics.
Age: Neonates have
reduced hepatic metabolism and renal excretion due to
relative organ immaturity. On the other hand,
elderly patients exhibit differences in absorption,
hepatic metabolism, renal clearance and volume of
distribution.
Genetic Factors:
Genetic polymorphism
affecting CYP2D6, CYP2C19, CYP2A6, CYP2C9, and N-acetyltransferase
result in significant inter-individual differences in
drug-metabolizing abilities (the drug of course must
be a substrate for one of the above cytochrome P450
isoforms)
Certain genetic
polymorphisms are associated with ethic groups.
For instance, 5%-10% of Caucasians are poor
metabolizers of CYP2D6 substrates. By contrast,
the frequency in Asian populations is about
1%-2%. On the other hand, the incidence of poor
metabolizers of CYP2C19 drugs is about 20% in Asian
populations, but only about 4% in Caucasian
populations.
Definition: genetic
polymorphism -- "Genetic polymorphism is a type
of variation in which individuals was sharply distinct
qualities co-exist as normal members of the
population" Ford, 1940.
Cytochrome P450
isoform naming conventions:
Review -- drug
biotransformation usually involves two phases,
phase I & phase II.
Phase I
reactions are classified typically as
oxidations, reductions, or hydrolysis of the
parent drug. Following phase I
reactions, the metabolites are typically more
polar (hydrophilic) which increases the
likelihood of their excretion by the
kidney. Phase I metabolic products may
be further metabolized
Phase II
reactions often use phase I metabolites can
catalyze the addition of other groups, e.g.
acetate, glucuronate, sulfate or glycine to
the polar groups present on the
intermediate. Following phase II
reactions, the resultant metabolite is
typically more readily excreted.
Most phase I
reactions are catalyzed by the cytochrome P450
system (CYP). This superfamily consists of
heme-containing isoenzymes which are mainly
localized in hepatocytes, specifically within the
membranes of the smooth endoplasmic reticulum.
The primary extrahepatic site containing CYP
isoforms would be enterocytes of the small
intestine.
The gene family
name is specified by an Arabic numeral, e.g. CYP3.
> 40% of sequence homology characterize CYP
isoforms within a family.
CYP families
are subdivided into subfamilies designated by
an upper case letter, it e.g. CYP3A .
Gene numbers
of individual enzymes are noted by a second
Arabic numeral following the subfamily letter,
e.g. CYP3A4.
CYP isoforms not
onlymetabolize many endogenous substances
including prostaglandins, lipids, fatty acids, and
steroid hormones but also metabolize (detoxify)
exogenous substances including drugs
Major CYP
isoforms responsible for drug metabolism include:CYP3A4,
CYP2D6, CYP2C9, CYP2C19, CYP1A2, CYP2E1 in in
certain cases CYP2A6 and CYP2D6
Important
enzymes for phase II reactions include
glutathione-S-transferases, UDP-glucuronosyl
transferases, sulfotransferases, N-acetyltransferases,
methyltransferases and acyltransferases.
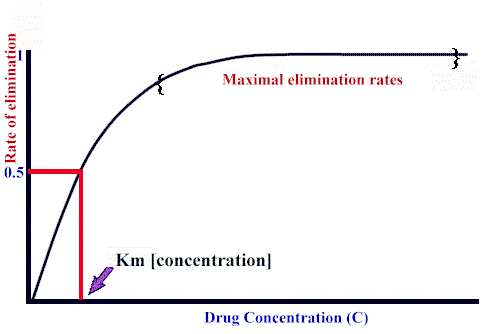
If blood flow to
the organ does not limit elimination, the
relationship between the elimination rate
and drug concentration,C, is:
Rate
of elimination = Vmax · C / (Km + C)
the form of this equation
is very similar to the Michaelis-Menten
description of enzyme kinetics. Here,
however:
Vmax
refers to maximum elimination
capacity
Km is
the drug concentration at which
the rate of elimination is 50% of
Vmax.
As expected from
the rectangular-hyperbolic shape
of the curve, at high drug
concentrations (compared to the Km),
dependency of elimination rate on
drug concentration decreases
significantly, approximating zero
order behavior. see below:
Holford, N. H.G. and Benet, L.Z.
Pharmacokinetics and Pharmacodynamics: Dose Selection and
the Time Course of Drug Action, in Basic and Clinical
Pharmacology, (Katzung, B. G., ed) Appleton-Lange, 1998,
pp 34-49.
Benet, Leslie Z, Kroetz, Deanna
L. and Sheiner, Lewis B The Dynamics of Drug Absorption,
Distribution and Elimination. In, Goodman and Gillman's
The Pharmacologial Basis of Therapeutics,(Hardman, J.G, Limbird, L.E,
Molinoff, P.B., Ruddon, R.W, and Gilman, A.G.,eds) The McGraw-Hill Companies, Inc.,1996, pp. 3-27
Stoelting, R.K.,
"Pharmacokinetics and Pharmacodynamics of Injected
and Inhaled Drugs", in Pharmacology and Physiology
in Anesthetic Practice, Lippincott-Raven Publishers,
1999, 1-17.
Half-life: (t1/2)
-- time required to decrease the amount
of drug in body by 1/2 during elimination
(or during a constant infusion).
Assumption:
single
body compartment size = volume of
distribution (Vd)
blood or
plasma considered in equilibrium
with total volume of distribution
t1/2
= (0.693 · Vd)/CL
t1/2
= (0.693)/kel
0.693 equals the
natural logarithm of two. {Since drug
elimination is an exponential process,
the time required for a twofold decreased
is proportional to ln(2)}.
kel
= km
+ kex;
where the elimination rate, kel
,constant is the sum of the rate
constants due to metabolism, km
, and excretion,kex.
Factors affecting t1/2:
disease
states-- affects volume of distribution
and clearance
decreased
Vd due to decreased renal
and skeletal muscle mass
(decreased digoxin tissue
binding)
resultant
increase in digoxin
half-life less than
expected based on renal
function change
example 2:
half-life of diazepam (Valium) increases
with age --
clearance
does not change
volume
of distribution changes
example 3:
half-life changes secondary to
changes in plasma protein
binding.
patients
with acute viral
hepatitis: half-life of Tolbutamide (Orinase)
decreases
(opposite of expected?)
Acute
viral hepatitis alters
plasma and tissue
drug-protein binding; the
disease does not change
volume of distribution
but increases total
clearance because more
free drug (not bound to
protein) is present.
Elimination halftime and anesthesia:
Elimination halftime is important in
estimating recovery from anesthetic drug administration.
In the case of IV administered agents, an
inconsistency between the elimination halftimes following a
single, bolus injection compared to continuous IV infusion,
has resulted in the development of an idea of referred to as
"context-sensitive or dependent" halftimes.
The definition of
"context-sensitive" halftimes is the length of
time required for the drug plasma concentration to fall
50% after continuous infusion is discontinued.
For IV anesthetic drug pharmacokinetics,
special problems exist because those significant
differences in individual drug requirements (up to 2-5
times) as a result of dose-plasma and plasma-effect
relationships
By contrast to the above special problems
associated with IV anesthetic drug pharmacokinetics and
variation between drugs, a similar problem does not exist for
the volatile agents were drug-effect relationships appear more
predictable.
Half-life:
Useful in estimating time
to steady-state: approximately 4
half-lives are required to reach about
94% of a new steady-state
Useful in estimating time
required for drug removal from the body
means for estimation of
appropriate dosing interval
Some drugs that
exhibit high extraction by the liver are
given orally. Some examples -- desipramine (Norpramin),
imipramine (Tofranil), meperidine (Demerol), propranolol (Inderal),
amitriptyline (Elavil, Endep), isoniazid (INH).
Some drugs which
have relatively low bioavailability are
not given orally because of concern of
metabolite toxicity -- lidocaine (Xylocaine) is an example (CNS
toxicity, convulsions)
High extraction
ratio drugs show interpatient
bioavailability variation because all of
sensitivity to:
hepatic function
blood flow
hepatic disease
(intrahepatic or extrahepatic
circulatory shunting)
Drugs poorly
extracted by the liver:
phenytoin (Dilantin)
diazepam (Valium)
digitoxin (Crystodigin)
chlorpropamide (Diabinese)
theophylline
Tolbutamide (Orinase)
warfarin (Coumadin)
Avoiding the first-pass effect:
sublingual (e.g.
nitroglycerin)-- direct access to
systemic circulation
transdermal
use of suppositories in
the lower rectum {if
suppositories move upward,
absorption may occur through the
superior hemorrhoidal veins,
which lead to the liver}
inhalation: first-pass
pulmonary loss by excretion or
metabolism may occur.
Holford, N. H.G. and Benet, L.Z.
Pharmacokinetics and Pharmacodynamics: Dose Selection and
the Time Course of Drug Action, in Basic and Clinical
Pharmacology, (Katzung, B. G., ed) Appleton-Lange, 1998,
pp 34-49.
Benet, Leslie Z, Kroetz, Deanna
L. and Sheiner, Lewis B The Dynamics of Drug Absorption,
Distribution and Elimination. In, Goodman and Gillman's
The Pharmacologial Basis of Therapeutics,(Hardman, J.G, Limbird, L.E,
Molinoff, P.B., Ruddon, R.W, and Gilman, A.G.,eds) TheMcGraw-Hill Companies, Inc.,1996, pp. 3-27