|
|
|
|
|
|
|
Nursing Pharmacology Chapter 33-34: Anticancer Drugs
|
|
|
|
|
|
|
Antimetabolites
Pyrimidine Analogues continued:
5-fluorouracil (Adrucil) may be administered directly as 5-FU or can be made available by administration of the prodrug, capecitabine (Xeloda).1
Capecitabine is converted to 5-FU.
Before 5-FU can exhibit cytotoxic action 5-FU requires enzyme catalyzed conversion to another form.1
Other reactions result in formation of the deoxy derivative FdUMP, 5-fluorodeoxyuridylate which inhibits thymidylate synthase blocking synthesis of deoxythymidine triphosphate (dTTP).1
dTTP is a required DNA constituent.
The complex consisting of a folate cofactor, 5,10-methylene FH4 and 5-fluorodeoxyuridylate (FdUMP) binds thymidylate synthase.
This complex does not allow thymidylate synthesis because the increased ability of the fluorine carbon bond (FdUMP) is sufficient to stop the reaction.1
In some circumstances, malignant cells appear to have inadequate concentrations of 5, 10-methylene FH4 thus limiting the concentration of the inhibited ternary complex associated with thymidylate synthase.1
![]() Improved
responsiveness to 5-fluorouracil may be obtained in these circumstances by
adding leucovorin, a source of exogenous folate.
Improved
responsiveness to 5-fluorouracil may be obtained in these circumstances by
adding leucovorin, a source of exogenous folate.
Other approaches have been employed to enhance 5-FU cytotoxicity.
Some approaches involve adding other drugs, for example methotrexate.
![]() Methotrexate
through purine synthesis inhibition increases 5-FU activation and improves
its antitumor efficacy.
Methotrexate
through purine synthesis inhibition increases 5-FU activation and improves
its antitumor efficacy.
For tumors of the "upper aerodigestive" tract, 5-FU in combination with cisplatin has proven helpful.1
5-FU along with leukovorin oxaliplatin is frequently employed for management of metastatic colorectal cancer (FOLFOX: folinic acid (FOL) + fluorouracil (F) + oxaliplatin (OX))1,8
Another combination involves adding irinotecan (FOLFIRINOX), a combination helpful in treating patients with colorectal or pancreatic cancer.1
![]() Fluoropyrimidines
have enhanced effects when combined with radiation.
Fluoropyrimidines
have enhanced effects when combined with radiation.
For example, 5-FU in combination with irradiation may be curative in patients with anal cancer and appears to improve local control of disease in patients with pancreatic, head and neck, cervical, rectal and gastroesophageal cancer.1
![]() 5-fluorouracil
is incorporated into both RNA and DNA instead of normal, physiological
thymidine triphosphate (TTP).
5-fluorouracil
is incorporated into both RNA and DNA instead of normal, physiological
thymidine triphosphate (TTP).
A cellular response to 5-FU incorporation is activation of excision-repair systems.
As a consequence, DNA strand breakage may occur since DNA repair requires physiological, thymidine triphosphate, unavailable due to thymidylate synthase inhibition.
RNA processing and function is adversely affected following 5-FU incorporation into RNA.
Abnormal RNA processing and function contributes to 5-FU cytotoxicity.1
Absorption, Distribution, Biotransformation, Excretion:
Following oral administration of 5-fluorouracil, 5-FU absorption tends to be unpredictable and sometimes incomplete.1
As a result, this agent is administered parenterally.3
Due to the short half-life of this agent (10-15 minutes), infusion is typically preferred over bolus administration.3
![]() Much of the 5-FU
administered dose, about 80-85%, is catabolized by a reaction catalyzed by
the enzyme dihydropyrimidine dehydrogenase (DPD).
Much of the 5-FU
administered dose, about 80-85%, is catabolized by a reaction catalyzed by
the enzyme dihydropyrimidine dehydrogenase (DPD).
This enzyme is encoded in humans by the DPYD gene in the enzyme is both the initial and rate-limiting step in pyrimidine catabolism.9
In addition to 5-fluorouracil degradation, the enzyme catalyzes degradation of tegafur (Tetrahydrofuranyl-5-fluorouracil), a 5-FU prodrug.
![]() Inherited
deficiencies of DPD results in increased 5-FU sensitivity.1,6
Inherited
deficiencies of DPD results in increased 5-FU sensitivity.1,6
Several laboratory tests are available to identify DPD deficiencies.
![]() Patients who
have deficiencies in dihydropyridines dehydrogenase (DPD) do not exhibit
this phenotype until challenged with 5-FU.6
Patients who
have deficiencies in dihydropyridines dehydrogenase (DPD) do not exhibit
this phenotype until challenged with 5-FU.6
Should patients respond to 5-FU or other fluoropyrimidine anticancer agents with excessive or severe toxicity, DPD deficiency must be considered. DPD mutations, however, appear insufficient to account fully for observed instances of excessive 5-FU toxicity.
As many as 50% of patients experiencing 5-FU toxicities do not exhibit DPD gene alterations.6
Furthermore, patients with normal DPD enzyme activity sometimes exhibit high 5-FU plasma levels which manifest as increased drug toxicities.
Although specialized laboratory tests using peripheral blood mononuclear cells can screen for DPD enzyme activity, these tests are not considered routine.
Routine phenotypic and genotypic DPD deficiency screenings prior to 5-FU treatment are likely not yet available.6
DPD deficiencies can result in increased toxicity risk for those patients receiving 5-FU chemotherapy.9
The DPD deficiency is autosomal recessive and this pharmacogenetic finding manifests as either complete or partial DPD enzyme deficiency may be noted in up to 5% of cancer patients.3
![]() The
increase in toxicity due to DPD deficiency may be quite severe, manifesting
as:
The
increase in toxicity due to DPD deficiency may be quite severe, manifesting
as:
Myelosuppression
G.I. toxicity (diarrhea and/or mucositis) and
Neurotoxicity.
These three toxic responses may be described as the "classic triad.3
About 5%-10% of a single 5-FU intravenous dose is recoverable as the parent compound in the urine.
5-FU partitions into the cerebrospinal fluid (CSF) only to a minimal degree.1
Resistance Mechanisms:
![]() The most
frequently identified resistance mechanism to the action of 5-FU is a change
in its target, the enzyme thymidylate synthase (TS).6
The most
frequently identified resistance mechanism to the action of 5-FU is a change
in its target, the enzyme thymidylate synthase (TS).6
Sensitivity to 5-fluorouracil-mediated cytotoxicity is strongly associated with thymidylate synthase enzymatic activity and the thymidylate synthase protein levels.
Thymidylate synthase levels are under tight autoregulatory control manifest by unbound enzyme directly modulating (inhibiting) its own mRNA translation.1
This system allows for rapid thymidylate synthase activity/level changes required for cell division.
Upon binding of thymidylate synthase to fluorodeoxyuridine monophosphate (FdUMP) translation inhibition is decreased with free thymidylate synthase increasing.
It is possible that thymidylate synthase autoregulation is an important contributing mechanism through which cancer cells become insensitive to 5-FU effects.1
Tumors that exhibit higher levels of TS appear more resistant to 5-FU cytotoxicity.6
Specific changes or mutations in thymidylate synthase have been associated with decreased binding affinity for the 5-FU metabolite, fluorodeoxyuridine monophosphate (FdUMP).
Abnormalities in enzymes that are required for cytotoxic 5-FU metabolite formation may also confer relative resistance.
Such abnormalities include: decreased enzyme protein expression along with or in addition to reduced enzymatic activity (enzyme activity per enzyme active site).
Fluoropyrimidine resistance has been correlated with increased expression of the degrading enzyme (the catabolic enzyme dihydropyrimidine dehydrogenase (DPD)).
Reduced expression of mismatch repair enzymes appear also associated with fluoropyrimidine resistance.
In the clinical setting, the relative contribution of the above factors to 5-FU cellular cytotoxicity resistance remains to be elucidated.6
|
Principle side effects associated with 5-FU administration are:
Mucositis
Diarrhea, and
Myelosuppression.
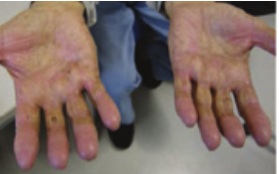
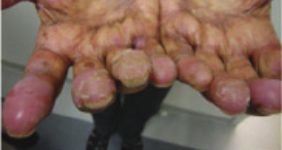
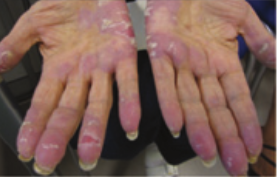
Dermatologic hand-foot syndrome (HFS) is more likely noted with infusional 5-FU administration.6
|
|
|
|
|
|
Acute neurological presentations have been described as including cerebellar ataxia somnolence and upper motor signs.6
Coronary vasospasm rarely can be induced following 5-FU treatment.
Cardiac toxicity is more likely associated with infusional compared to bolus 5-FU administration.6
5-fluorouracil has a number of important therapeutic uses as anticancer drugs.
These uses include:
Colon cancer
Breast cancer
Anal cancer
Gastroesophageal cancer
Hepatocellular cancer
Ppancreatic cancer, and
Head and neck cancer.
The method of administration varies as does the dosage depending on the particular neoplasm needing treatment. 5-fluorouracil can also be combined with other anticancer drugs.6
Floxuridine (5-fluorodeoxyuridine) is a precursor converted directly to FdUMP, catalyzed by the enzyme thymidine kinase.1
This agent may be used for continuous intrahepatic artery infusion for treating patients with metastatic colon cancer or after resection of hepatic metastatic disease.1
Floxuridine is one of a number of drugs which have been used also for intraperitoneal chemotherapy for treating gastric cancer (stomach cancer).11
Clinical trials have investigated efficacy of 5-fluorouracil, floxuridine, mitomycin C, and cisplatin.
Also, intraoperative intraperitoneal hyperthermic chemotherapy normal thermic intraoperative chemotherapy provided at the end of the operation, early postoperative intraperitoneal (normothermic) chemotherapy, or delay postoperative intraperitoneal (normothermic) chemotherapy have been evaluated.
Hyperthermic intraoperative peritoneal chemotherapy with or without early postoperative intraperitoneal (normothermic) chemotherapy treatment may be associated with some modest survival benefit as well as associate with elevated complications.11
Liver metastatic lesions derive perfusion from hepatic arterial sources.12
Floxuridine given via hepatic arterial infusion exposes these tumors to very high-dose chemotherapy.
![]() Adjuvant and
neoadjuvant hepatic arterial infusion chemotherapy have been associated with
notable benefits.
Adjuvant and
neoadjuvant hepatic arterial infusion chemotherapy have been associated with
notable benefits.
Floxuridine in particular exhibits favorable response rates for those patients refractory to contemporary chemotherapeutic drugs.12
![]() Patients
who receive hepatic arterial infusion chemotherapy must be closely watched
for signs of biliary cirrhosis.
Patients
who receive hepatic arterial infusion chemotherapy must be closely watched
for signs of biliary cirrhosis.
Cholangiocytes of the biliary tree receive blood supply from the hepatic arterial system; consequently, extended chemotherapy arterial infusion can induce biliary strictures.
This risk may be reduced if dexamethasone is administered along with floxuridine.12
![]() Response rates
following floxuridine intrahepatic infusion (about 50%) is about two times
that observed following intravenous administration.1
Response rates
following floxuridine intrahepatic infusion (about 50%) is about two times
that observed following intravenous administration.1
Two to three weeks of intrahepatic arterial infusion appear to cause limited systemic toxicity.
![]() Multiple
cycles of treatment, however, risk biliary cirrhosis, as described above.1
Multiple
cycles of treatment, however, risk biliary cirrhosis, as described above.1
Stomatitis or diarrhea, earliest occurrences of toxicity, is a basis for treatment discontinuation since greatest bone marrow suppression and GI toxicity are not observed until one to two weeks after treatment initiation.
|
|
|
|
|
|
|
|