|
|
|
|
|
|
|
Nursing Pharmacology Chapter 33-34: Anticancer Drugs
|
|
|
|
|
|
|
Hormonal Anticancer Drugs:
Luteinizing hormone releasing hormone agonist (LHRH):
Overview:6
In 1941 experiments demonstrated that reducing serum testosterone (by castration or estrogen treatment) induced regression of metastatic prostate cancer.6
Such approaches were designated as Androgen Deprivation Therapy (ADT).
![]() ADT
continues to be an element in prostate cancer therapy.
ADT
continues to be an element in prostate cancer therapy.
Occasionally, testicular surgery (bilateral orchiectomy) is performed to prevent testosterone treatment.
![]() Psychological as well as physical adverse effects follow from bilateral
orchiectomy which does retard disease advancement.
Psychological as well as physical adverse effects follow from bilateral
orchiectomy which does retard disease advancement.
Chemical castration which results in androgen deprivation represents the principal current therapeutic approach used to reduce serum testosterone to a level at least < 50 ng/dL (or <20 ng/dl).6
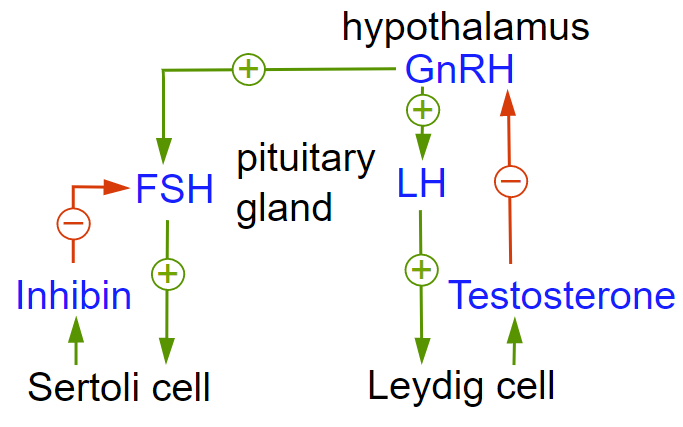
Currently ADT involves using GnRH analogs (Gonadotropin Receptor Hormone) which exhibit greater potency compared to natural GnRH.
|
These agents are used in treating prostate cancer and breast cancer.
![]() These
analogs bind to GnRH receptors (GnRH-R) in the pituitary resulting in an
initial increase in Lutinizing Hormone (LH) and Follicle
Stimulating Hormone (FSH) and testosterone.
These
analogs bind to GnRH receptors (GnRH-R) in the pituitary resulting in an
initial increase in Lutinizing Hormone (LH) and Follicle
Stimulating Hormone (FSH) and testosterone.
LH increases testosterone production by Leydig cells (testicular interstitial cells).
FSH increases testicular growth and production of androgen-binding protein by Sertoli cells required for supporting sperm cell maturation.
This initial "androgen surge" is associated with the first 3-7 days of treatment.
The initial increase (surge) in agonist activity associated with GnRH analog administration may result in a "flare" response due to elevated androgen levels.6.
This clinical flare may be associated with:
Elevated prostate volume
Spinal cord compression and
Bone pain.
Although no longer common practice, antiandrogen administration has been used to block flare symptoms.
Eamples of antiandrogens used for this purpose include flutamide or bicalutamide.6,3
However, the continuous activation of GnRH receptors causes gonatotroph cellular desensitization as well as reduced GnRH-R expression.
This response causes a reduction in Leutinizing-Hormone (LH) synthesis, thus halting testicular steroidogenesis.
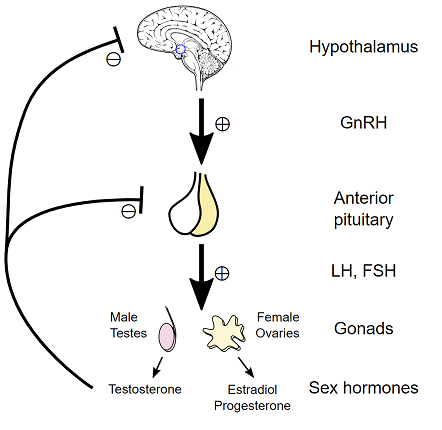
In summary, gonadal androgen synthesis is suppressed due to feedback inhibition of the Hypothalamic-Pituitary-Gonadal axis.6
|
|
|
|
|
|
The analogs, goserelin and leuprolide are given at monthly intervals, as intramuscular depot preparations.
This route of administration is considered preferable since parenteral administration results in rapid clearance of the analogs.3
![]() Goserelin (Zoladex
and others, GOE se rel in))
Goserelin (Zoladex
and others, GOE se rel in))
GnRH synthetic agonists [goserelin, Zoladex; triptorelin, Trelstar; leuprolide, Lupron], are used in treating prostate cancer.6
In that clinical setting, these agents are administered at high-dose as constant release formulation.
Depot formulations for goserelin (as well as for the other approved GnRH agonists) allow for durations of action extending from 1 to 6 months.6
Absorption, Distribution, Biotransformaion, Excretion:2
![]() GnRH
synthetic agonists [goserelin, Zoladex; triptorelin, Trelstar;
leuprolide, Lupron], are used in treating prostate cancer.
GnRH
synthetic agonists [goserelin, Zoladex; triptorelin, Trelstar;
leuprolide, Lupron], are used in treating prostate cancer.
In that clinical setting, these agents are administered at high-dose as constant release formulations.
As shown above, GnRH agonists or peptides, similar in structure to the naturally occurring hormone GnRH.
Goserelin exhibits a modification with respect to the naturally occurring GnRH in that a d-serine is altered with a t-butyl group. See above:
Depot formulations for goserelin (as well as for the other approved GnRH agonists) allow for durations of action extending from 1 to 6 months.
Goserelin administration (3.6 mg, depot route of administration) results in an early, short-duration goserelin peak (about 8 hours following administration).
A more prolonged peak is noted about two weeks following administration.
Different serum profiles are associated with the amount of depot drug.
For example, a 10.8 mg depot administration is associated with a decrease in plasma level with a small secondary peak at five weeks.2
Metabolism involves hepatic hydrolysis.2
Elimination of goserelin is mainly by renal excretion with over 90% of the agent found in the urine.
Most of the drug is excreted rapidly with about 75% excreted during the first 12 hours.
In the urine, about 20% of the administered drug is excreted unchanged with the remaining metabolic hydrolytic metabolic products constituting the remainder.
Goserelin clearance is reduced in this setting of renal disease and in that case an increase in drug half-life is noted.
By contrast, hepatic impairment has relatively minor effects on goserelin pharmacokinetics.
Importantly, due to the "white therapeutic window" of goserelin, renal or hepatic insufficiency does not require alteration in patient dosing. 2
![]() In 1941
experiments demonstrated that reducing serum testosterone (by castration
or estrogen treatment) induced regression of metastatic prostate cancer.6
In 1941
experiments demonstrated that reducing serum testosterone (by castration
or estrogen treatment) induced regression of metastatic prostate cancer.6
These analogs bind to GnRH receptors (GnRH-R) in the pituitary resulting in an initial increase in Lutinizing Hormone (LH) and Follicle Stimulating Hormone (FSH) and testosterone.
LH increases testosterone production by Leydig cells (testicular interstitial cells).
FSH increases testicular growth and production of androgen-binding protein by Sertoli cells required for supporting sperm cell maturation.
![]() This
initial "androgen surge" is associated with the first 3-7 days of
treatment.
This
initial "androgen surge" is associated with the first 3-7 days of
treatment.
The initial increase (surge) in agonist activity associated with GnRH analog administration may result in a "flare" response due to elevated androgen levels.6.
This clinical flare may be associated with:
Elevated prostate volume
Spinal cord compression and
Bone pain.
Although no longer common practice, antiandrogen administration has been used to block flare symptoms.
Examples of antiandrogens used for this purpose include flutamide or bicalutamide.6,3
![]() However, the continuous activation of GnRH receptors causes
gonatotroph cellular desensitization as well as reduced GnRH-R
expression.
However, the continuous activation of GnRH receptors causes
gonatotroph cellular desensitization as well as reduced GnRH-R
expression.
This response causes a reduction in Leutinizing-Hormone (LH) synthesis, thus halting testicular steroidogenesis.
In summary, gonadal androgen synthesis is suppressed due to feedback inhibition of the Hypothalamic-Pituitary-Gonadal axis.6
Goserelin is administered for palliative treatment of advanced prostate cancer (carcinoma of the prostrate) as well as for management of locally confined prostate cancer in combination with other therapeutic approaches.5
Other labeled indications include palliative treatment of advanced breast cancer, endometrial thinning, and endometriosis.5
![]() Side-Effects/
Adverse Effects:9
Side-Effects/
Adverse Effects:9
Numerous side effects associated with goserelin administration have been reported.
Some of these include:
Hot flashes
Impotence
Reduced frequency of erections.
Since this agent may be used to treat prostate cancer symptoms as well as, in women, breast cancer or endometriosis a broad range of gender-dependent side effects may be expected.9
GnRH synthetic agonists [goserelin, Zoladex; triptorelin, Trelstar; leuprolide, Lupron], are used in treating prostate cancer.6
Metastatic prostate cancer may be addressed by androgen deprivation.
For example, orchiectomy results in about an 80% response rate in patients.7
Instead of orchiectomy, testicular androgen suppression can be induced by agents such as leuprolide, goserelin (above) and others.
Furthermore, principal hormonal management in prostate cancer could typically involve orchiectomy or leuprolide (or related agents), but not both together.
The GnRH synthetic analogs are administered at high-dose as constant release formulation.
Depot formulations for luprolide (as well as for the other approved GnRH agonists) allow for durations of action extending from 1 to 6 months.6
![]() Leuprolide (Lupron) is not active when administered orally and is
therefore typically administered subcutaneously or more likely, as a
monthly intramuscular depot injection.6
Leuprolide (Lupron) is not active when administered orally and is
therefore typically administered subcutaneously or more likely, as a
monthly intramuscular depot injection.6
This route of administration allows for a slow, constant release of drug.
Using a depot formulation of 3.75 mg, a peak plasma level (4.6-10.2 ng/ml) is obtained in about 4 hours.
Plasma concentrations becomes stable after about two days (0.3 ng/ml), maintaining that level for about a month.
The steady-state volume of distribution (Vd) is approximately 30 L.
This agent exhibits a terminal elimination half-time of about 3 hours.6
Protein binding is in the range of 45%.11
Metabolism: The principal metabolite is an inactive pentapeptide (M-1) as well as other smaller inactive peptides.
Leuprolide metabolism does not involve the cytochrome P450 system.13
Excretion: About 5% of the intact drug and major metabolite is recoverable in the urine.11
The smaller, inactive peptides of leuprolide are formed as a result of the action of the enzyme peptidase.12
![]() Mechanism of Action:
Mechanism of Action:
In 1941 experiments demonstrated that reducing serum testosterone (by castration or estrogen treatment) induced regression of metastatic prostate cancer.6
These analogs bind to GnRH receptors (GnRH-R) in the pituitary resulting in an initial increase in Lutinizing Hormone (LH) and Follicle Stimulating Hormone (FSH) and testosterone.
LH increases testosterone production by Leydig cells (testicular interstitial cells).
FSH increases testicular growth and production of androgen-binding protein by Sertoli cells required for supporting sperm cell maturation.
![]() This
initial "androgen surge" is associated with the first 3-7 days of
treatment.
This
initial "androgen surge" is associated with the first 3-7 days of
treatment.
The initial increase (surge) in agonist activity associated with GnRH analog administration may result in a "flare" response due to elevated androgen levels.6.
This clinical flare may be associated with:
Elevated prostate volume
Spinal cord compression and
Bone pain.
Although no longer common practice, antiandrogen administration has been used to block flare symptoms.
Eamples of antiandrogens used for this purpose include flutamide or bicalutamide.6,3
However, the continuous activation of GnRH receptors causes gonatotroph cellular desensitization as well as reduced GnRH-R expression.
![]() This
response causes a reduction in Leutinizing-Hormone (LH) synthesis, thus
halting testicular steroidogenesis.
This
response causes a reduction in Leutinizing-Hormone (LH) synthesis, thus
halting testicular steroidogenesis.
In summary, gonadal androgen synthesis is suppressed due to feedback inhibition of the Hypothalamic-Pituitary-Gonadal axis.6
Leuprolide is administered for palliative treatment of advanced prostate cancer (carcinoma of the prostrate) as well as for management of locally confined prostate cancer in combination with other therapeutic approaches.5
Numerous side effects associated with leuprolide administration have been reported.
Some of these include:
Hot flashes
Impotence
Reduced frequency of erections.
Since this agent may be used to treat prostate cancer symptoms as well as, in women, breast cancer or endometriosis a broad range of gender-dependent side effects may be expected.9
Triptorelin (Decapeptyl, Gonapeptyl)14
Triptorelin is a synthetic GnRH agonist available as either an acetate or a pamolate salt.
Triptorelin is an example of a GnRH synthetic agonist [along with goserelin, Zoladex and leuprolide, Lupron], used in treating prostate cancer.6
Metastatic prostate cancer may be addressed by androgen deprivation. For example, orchiectomy results in about an 80% response rate in patients.7
Instead of orchiectomy, testicular androgen suppression can be induced by agents such as triptorelin (Decapeptyl, Gonapeptyl, Trelstar (pamolate formulation)) and others.
Triptorelin pamolate (Trelstar) is a decapeptide, (pGlu-His-Trp-Ser-Tyr-D-Trp-Leu-Arg-Pro-Gly-NH2).12
Absorption, Distribution, Biotransformation, Excretion:
Triptorelin (pamolate) is available for intramuscular depot injection (buttock) at several doses corresponding to differing dosage intervals.6
In vitro, triptorelin exhibits more biological activity compared to native GnRH (by about a factor of 100).
Triptorelin, when administered as a subcutaneous depot formulation achieves peak serum levels in about 2 hours, a timeframe unaffected by which dosage is chosen (3.75 mg, 11.25 mg or 22.5 mg).
The volume of distribution (Vd) for triptorelin is about 30 L.6
Triptorelin is not associated with protein binding.14
Metabolic pathways which may be involved in triptorelin degradation remain to be identified but appear unlikely to be cytochrome P450 dependent.6,14
Triptorelin is eliminated by both hepatic and renal pathways with about 40% of an administered dose recovered intact in the urine.
In patients with hepatic dysfunction, the percentage of intact triptorelin recovered in the urine is increased by about 50%.6
In 1941 experiments demonstrated that reducing serum testosterone (by castration or estrogen treatment) induced regression of metastatic prostate cancer.6
These analogs bind to GnRH receptors (GnRH-R) in the pituitary resulting in an initial increase in Lutinizing Hormone (LH) and Follicle Stimulating Hormone (FSH) and testosterone.
LH increases testosterone production by Leydig cells (testicular interstitial cells).
FSH increases testicular growth and production of androgen-binding protein by Sertoli cells required for supporting sperm cell maturation.
![]() This
initial "androgen surge" is associated with the first 3-7 days of
treatment.
This
initial "androgen surge" is associated with the first 3-7 days of
treatment.
The initial increase (surge) in agonist activity associated with GnRH analog administration may result in a "flare" response due to elevated androgen levels.6.
This clinical flare may be associated with:
Elevated prostate volume
Spinal cord compression and
Bone pain.
Although no longer common practice, antiandrogen administration has been used to block flare symptoms.
Eamples of antiandrogens used for this purpose include flutamide or bicalutamide.6,3
However, the continuous activation of GnRH receptors causes gonatotroph cellular desensitization as well as reduced GnRH-R expression.
![]() This
response causes a reduction in Leutinizing-Hormone (LH) synthesis, thus
halting testicular steroidogenesis.
This
response causes a reduction in Leutinizing-Hormone (LH) synthesis, thus
halting testicular steroidogenesis.
In summary, gonadal androgen synthesis is suppressed due to feedback inhibition of the Hypothalamic-Pituitary-Gonadal axis.6
Triptorelin is administered for palliative treatment of advanced prostate cancer (carcinoma of the prostrate) as well as for management of locally confined prostate cancer in combination with other therapeutic approaches.5
Numerous side effects associated with triptorelin administration have been reported.
Some of these include:
Hot flashes
Impotence
Reduced frequency of erections.
Since this agent may be used to treat prostate cancer symptoms as well as, in women, breast cancer or endometriosis a broad range of gender-dependent side effects may be expected.9
|
|
|
|
|
|
|