![]()
![]()
|
|
|
Anesthesia Pharmacology Chapter 29: Clinical Correlation: Pulmonary Hypertension
![]()
![]()
|
|
|
|
|
|
2Possible causes:
The general suggestion has been that their use a generalized vascular hyper-reactivity, such that vessels constrict in response to certain factors. These factors may be external or internal. Along these lines, patients with pronounced disease appear more likely to develop primary PH.
Factors include: diet suppressants, cocaine, HIV, and pregnancy. These factors promote in susceptible patients pulmonary artery narrowing.
Genetics: about 6%-10% of cases exhibit familial linkage.
3Patient presentation:
Dyspnea, precordial discomfort, angina
Physical examination may reveal cyanosis and cardiomegaly
2Disease progression:
The initial insult may be in injury to the vascular endothelial cells
Subsequent to this injury, the intrinsic interrelationships between the endothelial cells and the underlying vascular smooth muscle may be changed. The direction of the change is to cause increased smooth muscle contraction.
As the disease progresses, vessel wall muscle cell proliferation causes thickening. Not only do regions possessing muscle cells normally thickening, but also muscle cells appear in regions not typically exhibiting vascular smooth muscle
Vascular stiffness with scarring and fibrosis accompanies thickening; vessels may become completely occluded; within smaller arteries, there is an increased likelihood for thromboembolism.
A secondary consequence is that the right ventricular myocardial muscle mass increases in order to pump against this increasing vascular resistance. Ultimately, following cardiac dilatation hypertrophy, right ventricular failure may occur.
Pulmonary hypertension (PH): Overview
1Clinical consequences: significant morbidity/mortality associated with right-sided cardiac failure and sudden death
Problematic diagnosis at least initially; however, acute PH is difficult to manage as it is associated with significant mortality.
The most successful treatment management is associated with early diagnosis allowing intervention preceding significant damage to the pulmonary vasculature
Most common cause of acute pulmonary hypertension:
pulmonary thromboembolism [perhaps up to 15% of hospital admitted patients have complications of pulmonary embolism]
|
|
|
|
|
|
|
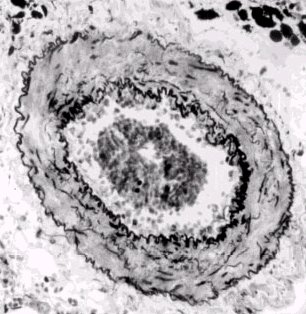
Pulmonary embolus-1: Summary of the above case
|
1Pathophysiological comments:
There are key interrelationships between right ventricular function and pulmonary circulation properties
Normal physiology: normally minimal pulmonary vascular bed resistance to blood flow
Pathophysiology: Pulmonary hypertension occurs because of either:
pulmonary vascular vasoconstriction
OR increase in pulmonary blood flow
These pathophysiological changes may be referable to pulmonary parenchymal abnormalities, myocardial abnormalities, and/or pulmonary vascular pathologies.
1Normal pulmonary circulation physiology:
The pulmonary circulation is a high-volume low pressure system which is in contrast to the systemic circulation.
Pulmonary artery mean pressure is about 12 mm Hg. The left atrial mean pressure ranges from 2 to 6 mm Hg; therefore, the normal pulmonary circulatory pressure gradient is only about 6 mm Hg. For comparison, the pressure gradient between the left ventricle and right atrium ranges from 80-100 mm Hg [for cardiac output of about 5 liters per minute]. The systemic vascular bed has a resistance to blood flow of about 10 times that observed for the pulmonary circulatory system.
For the high flow low pressure pulmonary system, only small differences in either right ventricular volume or pressure load can alter function in an important manner.
By definition, pulmonary hypertension occurs if the resting pressure is > 20 mm Hg or the resting pulmonary artery systolic pressure is > 30 mm Hg.
1Factors that influence pulmonary vasomotor tone:
Histamine
norepinephrine
angiotensin II
serotonin
arachidonic acid metabolites
lipoxygenase products
acetylcholine will induce a temporary vasodilation in patients with elevated pulmonary vascular resistance; moreover, this effect is probably mediated by release of endothelial-derived relaxing factor (nitric oxide) from the vascular endothelium.
1Hypoxic pulmonary vasoconstriction:
Pulmonary vasoconstriction may be induced as part of a mechanism to balance capillary perfusion with alveolar ventilation.
These vascular tone adjustments occur at the level of small pulmonary arteries and arterioles; however, the underlying mechanism remains to be elucidated definitively. It is possible that local hypoxia causes local histamine release which results in H1-receptor-mediated vasoconstriction.
Although vascular endothelium releases the smooth muscle relaxing chemical nitric oxide (NO), endothelial cells also release a vasoconstricting peptide endothelin. Probably the balance between nitric oxide and endothelin release is important in setting local pulmonary vasomotor tone.
|
|
|
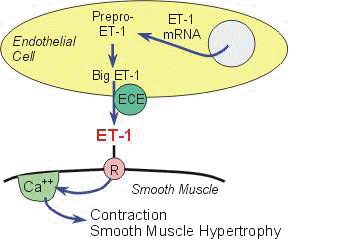
3ET-1 (endothelin-1) is secreted as "big-endothelin-1", which is a 38 amino acid peptide that is converted to ET-1 by the action of a protease [ET-1 converting enzyme-1, ECE-1].
There are two subtypes of ECE-1 which exhibit different anatomical distributions. ECE-1A is an endothelial cell product and ECE-1B is found on smooth muscle cells.
3There are two principal effects of ET-1: vasoconstriction and a mitogenic effect on smooth muscle [i.e. a stimulant of cellular proliferation]. In evaluation of patients with pulmonary hypertension, there appears to be correlation between disease severity and the level of ET-1 molecular expression. Molecular biological methods demonstrated that the higher levels of ET-1 were associated with higher levels of ET-1 mRNA.
3That ET-1 levels are important in the pathophysiology of pulmonary hypertension is supported by the observation that ECE-1 inhibitors (that is chemicals which inhibits the converting enzyme which creates ET-1 from "big-endothelin-one") reduce pulmonary artery pressure.
3Endothelins Receptors: Three endothelin receptor subtypes have been noted (ET-A, ETB-1, ETB-2).
ET-A receptors appear localized mainly on smooth muscle cells;
ETB receptors appear mainly associated with endothelial cells. A complexity that must be noted is that under some circumstances ET-1 (endothelin-1) has been observed to cause vasodilation-probably by interacting with the ETB receptor subtype, whereas interacting with the ETA subtype causes vasoconstriction. The prominent effect however is the vasoconstrictive effect.
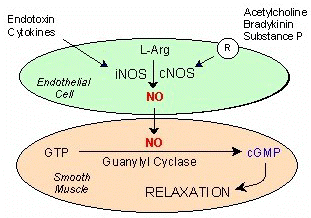
3,4The nitric oxide story:
Endothelial cells produce nitric oxide (NO,N=O) which is a vasodilator and vasodilates by activating guanylyl cyclase thus increasing cGMP.
cGMP can activate a GMP-dependent protein kinase, activate K+ channels, decrease IP3, and inhibit calcium entry into the vascular smooth muscle; these actions likely mediate vascular relaxation.5
NO also in may produce vasodilation by decreasing vasoconstrictor factor expression.
Therefore the amount of nitric oxide synthesis is of interest in view of its center role in potentially modulating pulmonary vasomotor tone in pulmonary hypertension.
4NO causes many biological effects including acting as a "bronchodilator, neurotransmitter, anticoagulant, anti-microbial agent, antithrombogenic factor, antiaggregatory factor, inhibitor of smooth muscle mitogenesis and a vasodilator"
Nitric oxide is synthesized from L-arginine (oxidative cleavage of the guanidino nitrogen) in a reaction catalyzed by nitric oxide synthase (NOS) and one requiring molecular oxygen and produces nitric oxide and L-citrulline.
NOS is a complex enzyme, depending upon five redox cofactors: NADPH, FAD, FMN, heme and tetrahydrobiopterin.
Furthermore there are three forms of NOS: a neuronal form (nNOS a.k.a.NOS-I),an inducible form (iNOS a.k.a. NOS II) and endothelial NOS or eNOS.
nNOS and eNOS are induced by calcium, whereas iNOS is activated by lipopolysaccharides, other inflammatory agents and cytokines.
iNOS catalyzes the production of the most nitric oxide
The significance of the level of NOS activity is in part due to the relationship between NOS activity and pulmonary hypertension disease severity.
eNOS levels in human disease correlate inversely with total pulmonary vascular resistance than pulmonary arteropathic severity.
By contrast, normal lungs stained intensely for eNOS in both pulmonary vascular endothelium and pulmonary epithelium.
A possible therapy for pulmonary hypertension might be therefore increasing the levels of nitric oxide or reducing the levels of ET-1
|
|
|
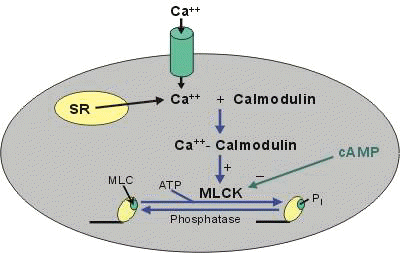
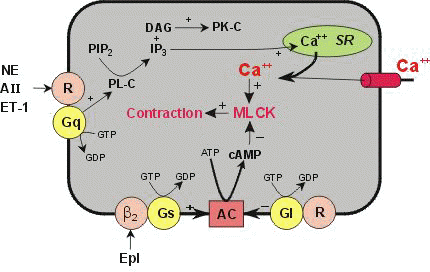
Smooth muscle vascular tone ultimately depends on activation of myosin light chain kinase with subsequent phosphorylation of myosin light chains. This step promotes interaction between actin and myosin. Myosin light chain kinase activation requires calcium complex with calmodulin. Therefore it is not surprising the calcium channel blocking drugs reduce the vasoconstrictive pulmonary response to hypoxemia since the reduction in intracellular calcium would limit myosin light chain kinase activation.
|
|
|
5Richard E. Klabunde, Ph.D.,Cardiovascular Physiology Web Resource used with permission
1Development of pulmonary hypertension
Right ventricular dysfunction may be caused by factors that also contribute to the development of pulmonary hypertension and include:
anatomical structural characteristics of the right ventricle
functional relationships between the right and left ventricle
factors that influence right ventricular coronary blood flow
1Primary pathophysiological consequence of PH: progressive attenuation of right ventricular function due to progressive increase in right ventricular afterload
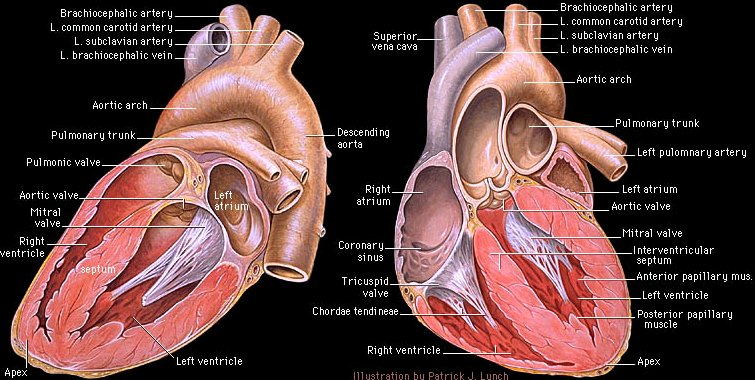
Anatomical considerations -- right vs. left ventricle: The right ventricle is relatively thin-walled, compliant and easily distended.
Also compared to the left ventricle, the right ventricle is considerably more sensitive to afterload increases, that is, small increases in ejection resistance leads to marked decreases in right ventricular stroke volume.
|
|
|
1Note in the image above the relatively thin right ventricular wall compared to the left ventricular wall. Furthermore, given the compliance of the right ventricle in the sensitivity of the right ventricle to small afterload increases, compensatory right ventricular wall tension increases may readily occur.
Such increases in wall tension are directly linked to increases in myocardial oxygen requirements, which is not met, may cause a deterioration right in right ventricular function.
1Anatomically, the heart must be described as two pumps operating in parallel and synchrony. Therefore if an output-mismatch occurs for even a short interval, vascular congestion will result -- either on the pulmonary side or systemically.
In accord with the difference in right vs. left ventricular muscle mass, the workload of the right ventricle is only about 15 percent of the left ventricle, despite the need to pump about the same cardiac output. This rough equivalence in cardiac output is only possible given the significantly reduced right vs. left afterload (resistance).
Increases in right ventricular afterload, even small increases, would be sufficient to shift the ventricular septum from right to left and thus impair left ventricular function. Additionally, septal shift (right to left) which would be associated with increased right ventricular end-diastolic volumes, impair left ventricular diastolic filling (increased filling pressures required to obtain a given diastolic left ventricular volume)
The above phenomena connect the increase in pulmonary artery pressure to a reduction left ventricular volume.
increased pulmonary artery pressure leads to increased right ventricular end diastolic volume
increased right end diastolic pressure shifts the intraventricular septum to the right which interferes with left ventricular function
reduce less ventricular function causes reduced cardiac output leading to increase pulmonary capillary wedge pressure
1The general rule is that coronary vascular perfusion occurs during diastole.
This assessment focuses on the coronary vasculature supplying the left side of the heart.
However the right side of the heart supplied by right ventricular coronary flow normally can occur both in systole and diastole, assuming normal, nonhypertrophied muscle and attendant low intracavity pressure.
Now consider what happens with right ventricular hypertrophy with higher intracavity pressures: The higher intracavity pressure tends to restrict coronary flow to diastole with its greater pressure gradient. Accordingly, the right side of the heart is much more sensitive to systemic hypotension in terms of getting adequate oxygen perfusion through coronary flow. Therefore, reduced coronary flow can further exacerbate right ventricular function secondary to hypotensive states and higher right ventricular intracavity pressures.
1Anatomical causes of pulmonary hypertension:
left ventricular dysfunction
left atrial lesions (e.g. ,mitral valve stenosis, mitral valve endocarditis, myxomas, thrombi)
pulmonary circulation lesions
right ventricular lesions
The above conditions promote long-term increases in pulmonary venous pressures which ultimately cause right ventricular hypertrophy, dilatation and ultimately right-sided pump failure.
Considering the consequences of left ventricular lesions: with inadequate left ventricular contractility or valvular dysfunction, elevation in left ventricular end diastolic volume will increase pulmonary venous pressure promoting pulmonary hypertension.
Factors responsible for left ventricular dysfunction include valvular dysfunction (mitral valve dysfunction), ischemic coronary vascular disease, as well as idiopathic/infectious cardiomyopathies. Again the effect on right ventricular performance will be indirect since these left ventricular lesions all increase the right-side afterload, i.e. right ventricular ejection resistance.
Obstructive disease: these lesions cause an increase pulmonary venous an arterial systemic pressure.
1Pulmonary hypertension in the clinical settings: acute vs. chronic presentations and pathologies
Pulmonary vascular resistance can be induced by many factors including hypoxia, acidosis, increase sympathetic tone as well as exogenous/endogenous vasoconstrictors such as thromboxane, catecholamines, and serotonin, hypercarbia and acidosis
Patients presenting perioperatively with significant pulmonary hypertension often will have a combination of both the chronic form within acute increase in pulmonary vascular resistance [as noted above, increase circulating catecholamines associated with perioperative stress might be sufficient to exacerbate underlying pulmonary hypertension]
Initial management perioperatively, may be centered on reversing at least the acute component. In considering a specific example, pulmonary vasoconstrictors or reduced endogenous nitric oxide from endothelial injury associated with cardiopulmonary bypass could explain increased right ventricular dysfunction after this procedure. Possibly inadequate interoperative protection of the right ventricle could be a contributing factor.
Chronic settings:
In chronic disease there has been long-term significant, fundamental changes in the pulmonary vasculature.
There is a fairly long list of contributing factors including primary PH, thromboembolic disease, vasculitis. Additionally parenchymal lung damage due to fibrosis or COPD promotes PH.
Abnormal ventilation such as obesity-hypoventilation syndrome or even sleep apnea may contribute to PH.
Early stage and then later stage considerations:
Increased pulmonary vascular resistance detected early can be managed with drugs. Unfortunately over time fundamental changes in the vasculature reduce pharmacological options. The vasculature changes include remodeling which is consistent with the mitogenic effects of ET-1.
As resistance to right ventricular ejection increases over time, the right ventricle is stressed and responds in a manner limited by limited compensatory reserves.
1Evaluation of right ventricular function
Since early identification of increased pulmonary vascular resistance allows effective pharmacological intervention, there is value in understanding initial clinical presentations however difficult to discern. Early presentations include dyspnea upon exertion, whereas more advanced pulmonary hypertension is associated with more serious clinical concerns including syncope, chest pain, or even sudden death. Clearly there is a great interest in identifying the disease early in the process begins with the physical examination.
1Physical examination: Factors consistent with pulmonary hypertension:
increase jugular venous distention
systolic murmur increasing in intensity during inspiration (consistent with tricuspid valve regurgitation)
pronounced second heart sound at the base of a heart -- due to forceful pulmonic valve closure
diastolic pulmonary valve murmur along less stern border
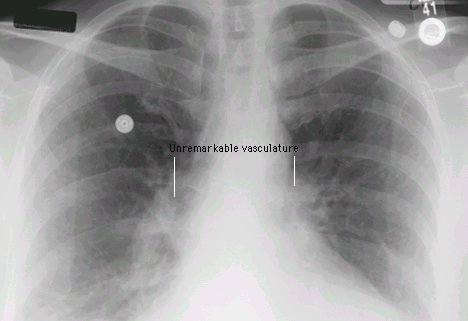
1Chest radiography:
Enlarged central, right, and left main pulmonary arteries in the medialstinal hilum.
Enlarged right ventricle may be observed in severe cases
Chest films may correlate with worsening symptoms.
1Electrocardiography (ECG):
Right ventricular enlargement may be sufficient to induce detectable ECG changes. These changes may not affect the basic cardiac rhythm; however, other changes may be discernible, including:
tall R wave and small S wave: lead V1 (R wave > 7 mm, S wave < 2 mm; R/S > 1)
tall S wave and small R wave and lead V5 or V6 (R/S < 1)
right axes deviation --QRS axis > 90 degrees
ST-segmental depression and T wave inversion in anterior leads (V1 through V4) indicative of significant right-sided heart dysfunction
right atrial enlargement would be indicated by P waves > 2.5 mm and leads II, III, and aVF
Note that traditional lead placement may make it difficult to detect early right ventricular dysfunctional indications; however, a new specially placement using the V4R lead in the fourth intercostal space in the mid-clavicular line has been shown to exhibit increased specificity insensitivity to correlations with other diagnostic methods such as radionuclide and echocardiographic standards for detecting right ventricular ischemia.
1Central venous and pulmonary artery pressures and thermal dilution
This method is now considered an inaccurate predictor of right ventricular end diastolic pressure and volume
Appropriate generalizations based on CVP hemodynamic patterns:
Decreased CVP is caused by decrease preload: appropriate therapy is volume administration (of course volume administration must proceed carefully in order to avoid further worsening of right ventricular function through distention)
Increased CVP is caused by decreased contractility which may require inotropic agents.
Thermal dilution methodologies may approximate right ventricular ejection fractions, although there are specific limitations that affect the accuracy of this method
1CT scan measurements:
inner endocardial wall identification
measurements of total ventricular stroke volume
epicardial surface outlines
wall thickness (ultrafast CT)
This technology can demonstrate increased right ventricular muscle mass in patients with PH
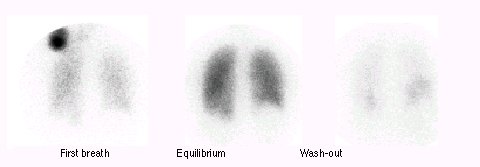
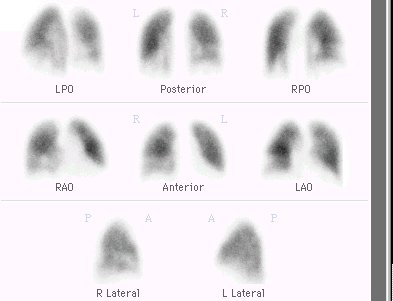
1Nuclear imaging:
Right ventricular and systolic pressure-volume relationships may be estimated using radionuclide ventriculogram; furthermore variations of right ventricular volume with varying loads can be determined
1Echocardiography:
Echocardiography has been an important technology in the assessment of right ventricular dimensions as a function of loads. Movement of the interventricular septum may also be resolved -- as noted earlier with increasing right ventricular pressures septal shift would be expected (right to left)
A number of important measurements ranging from the description of right atrial and ventricular dimensions to assessment of valvular regurgitation are available using electrocardiographic technologies.
1Afifi, S. and Hines, R., Seminars in Anesthesia, Perioperative Medicine and Pain, Vol 20, No 2 (June), 2001in: pp 118-132.9
2Division of Lung Diseases and Office of Prevention,
Education, and Control; Primary pulmonary hypertension; National Institutes of Health; National Heart, Lung, and Blood Institute, 1996;3Bousette, N, D'Orleans-Juste, P, Shennib,H, Giaid, A., Concise Review: The Role of Nitric Oxide and Endothelin-1 in Pulmonary Hypertension, Harrison's On-Line, McGraw-Hill, 2001
4Hart, CM: Nitric oxide in Adult Lung Disease, Chest 115:1407, 1999
5Richard E. Klabunde, Ph.D.,Cardiovascular Physiology Web Resource
![]()
![]()
|
|
|