|
|
|
Anesthesia Pharmacology: Antiarrhythmic Agents
|
|
|
|
|
|
|
![]() Class
I: Sodium Channel Blockers
Class
I: Sodium Channel Blockers
Sodium channel blocking antiarrhythmic drugs are classified as use-dependent in that they bind to open sodium channels.
Their effectiveness is therefore dependent upon the frequency of channel opening.
![]() There are three classes or types of sodium channel blockers:
There are three classes or types of sodium channel blockers:
Type Ia: prototype: quinidine gluconate (Quinaglute, Quinalan). Type Ia drugs slow the rate of AP rise and prolong ventricular effective refractory period.
Quinidine
Overview
Dextroisomer of quinine; quinidine gluconate (Quinaglute, Quinalan) also has antimalarial and antipyretic effects
Pharmacokinetics:
80%-90%: bound to plasma albumin
Rapid oral absorption; rapid attainment of peak blood levels (60-90 minutes)
Elimination half-life: 5-12 hours
IM injection, possible but not recommended due to injection site discomfort
IV administration: limited due to myocardial depression and peripheral vasodilation
Metabolism:
Hepatic: hydroxylation to inactive metabolites; followed by renal excretion
20% excreted unchanged in urine
Impaired hepatic/renal function: accumulation of quinidine and metabolites
Sensitive to enzyme induction by other agents, e.g.
Decreased quinidine blood levels with phenytoin, phenobarbital, rifampin
Mechanism of antiarrhythmic action-- primarily activated sodium channel blockade which results in:
Depression of ectopic pacemaker activity
Depression of conduction velocity
May convert a one-way conduction blockade to a two-way (bidirectional) block -- terminating reentry arrhythmias
Depression of excitability (particularly in partially depolarized tissue)
Recovery from sodium channel blockade is slower in depolarized tissue (compared to normal tissue):
This is the basis for relative selectivity of quinidine action in depolarized tissue compared to normal tissue, (i.e. lengthened refractory period, depressed conduction velocity, reduced excitability observed in depolarized tissue to greater extent the normal tissue)
Although classified as a sodium channel blocker, quinidine also blocks K+ channels.
Most antiarrhythmic agents have such multiple actions.
Effect on the ECG: QT interval lengthening
Basis: quinidine-mediated reduction in repolarizing outward potassium current
Result:
Longer action potential duration
Increased effective refractory period
Reduces reentry frequency; reduced rate in tachyarrhythmias
Sodium channel blockade results in:
An increased threshold
Decreased automaticity.
Used to manage nearly every form of arrhythmia especially acute and chronic supraventricular dysrhythmias
Ventricular tachycardia
Frequent indications:
Prevent recurrence of supraventricular tachyarrhythmias
Suppression ventricular premature contractions
Approximately 20% of patients with atrial fibrillation will convert to normal sinus rhythm following quinidine treatment Supraventricular tachyarrhythmia due to Wolff-Parkinson-White syndrome -- effective suppression by quinidine
Digitalization prior to quinidine administration:
Quinidine sulfate (Quinidex,Quinora)/quinidine gluconate (Quinaglute, Quinalan) may cause a paradoxical increase in ventricular response due to quinidine's vagolytic effect at the AV node
Quinidine's antimuscarinic action increases AV nodal throughput, allowing more SA nodal impulses to reach the ventricle.
Vagotonic effects on digitalis prevents this paradoxical increase by increasing vagal tone at AV node
Quinidine sulfate (Quinidex,Quinora) administration results in vagal inhibition (anti-muscarinic) and alpha-adrenergic receptor blockade.
Quinidine Side Effects
Cardiovascular--at (high) plasma concentrations (> 2ug/ml)
Prolongation (ECG) of PR interval, QRS complex, QT interval
Heart block likely with 50% increase in QRS complex duration (reduced dosage)
Quinidine syncope: may be caused by delayed intraventricular conduction, resulting in ventricular dysrhythmia
Patients with preexisting QT interval prolongation or evidence of existing A-V block (ECG): probably should not be treated with quinidine
Hypotension -- primarily following IV administration
Mechanism: peripheral vasodilation secondary to alpha-adrenergic receptor blockade
Increased hypotension risk associated with quinidine +verapamil treatment
Effects on heart rate:
Increase secondary to either quinidine's antimuscarinic effect and/or reflex increase in sympathetic activity
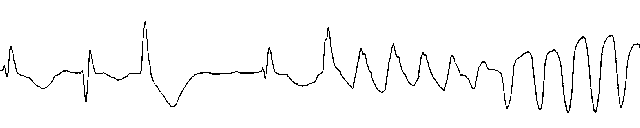
Quinidine is associated with Torsades de pointes, a ventricular arrhythmias associated with marked QT prolongation.
|
|
Torsades de pointes: Electrophysiological Features
Ventricular origin
Wide QRS complexes with multiple morphologies
Changing R - R intervals
Axis seems to twist about the isoelectric line
This potentially serious arrhythmia occurs in 2% - 8% if patients, even if they have a therapeutic or subtherapeutic quinidine blood level.
Other quinidine adverse effects include:
Cinchonism
Blurred vision, decreased hearing acuity, gastrointestinal upset,headaches and tinnitus.
Nausea, vomiting, diarrhea (30% frequency)
Drug-drug interaction:quinidine gluconate (Quinaglute, Quinalan)-digoxin (Lanoxin, Lanoxicaps)
Quinidine increases digoxin plasma concentration; may cause digitalis toxicity in patients taking digoxin or digitoxin
Effects on neuromuscular transmission:
Quinidine gluconate (Quinaglute, Quinalan) interferes with normal neuromuscular transmission; enhancing the effect of neuromuscular-blocking drugs
Recurrence of skeletal muscle paralysis postoperatively may be associated with quinidine administration
![]() Procainamide
Procainamide
Local anesthetic (procaine) analog
Long-term use avoided because of lupus-related side effect
Metabolism:
Elimination: renal excretion and hepatic metabolism; by contrast to procaine, procainamide is highly resistant to hydrolysis by plasma esterases.
40%-60% excreted unchanged (renal)
Renal dysfunction requires procainamide dosage reduction
Hepatic metabolism -- acetylation
Cardioactive metabolite: N-acetylprocainamide (NAPA);
NAPA accumulation may lead to Torsades de pointes
Quinidine and Procainamide similar: electrophysiological properties.
Possibly somewhat less effective in suppressing automaticity; possibly more effective in sodium channel blockade in depolarized cells
Useful in acute management of supraventricular and ventricular arrhythmias.
Drug of second choice for management of sustained ventricular arrhythmias (in the acute myocardial infarction setting)
Effective in suppression of premature ventricular contractions and paroxysmal ventricular tachycardia rapidly following IV administration
Most important difference compared quinidine: procainamide does not exhibit vagolytic (antimuscarinic) activity.
Procainamide is less likely to produce hypotension, unless following rapid IV infusion
Ganglionic-Blocking Activity
![]() Side
Effects/Toxicities
Side
Effects/Toxicities
Long term use is associated with side effects, including a drug-induced, reversible lupus erythematosus-like syndrome which occurs at a frequency of 25% to 50%.
Consists of serositis, arthralgia and arthritis
Occasionally: pluritis, pericarditis, parenchymal pulmonary disease
Rare: renal lupus
Vasculitis not typically present (unlike systemic lupus erythematosus)
Positive antinuclear antibody test is common; symptoms disappear upon drug discontinuation
In slow acetylators the procainamide-induced lupus syndrome occurs more frequently and earlier in therapy than in rapid acetylators.
Nausea, Vomiting -- most common early, noncardiac complication
Hondeghem, L.M. and Roden, D.M., "Agents Used in Cardiac Arrhythmias", in Basic and Clinical Pharmacology, Katzung, B.G., editor, Appleton and Lange, 1998, pp 216-241; Stoelting, R.K., "Cardiac Antidysrhythmic Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, 331-343
![]() Disopyramide (Norpace)
Disopyramide (Norpace)
Overview:
Very similar to quinidine gluconate (Quinaglute, Quinalan)
Greater antimuscarinic effects (in management of atrial flutter and fibrillation, pre-treatment with a drug that reduces AV conduction velocity is required)
Approved use (USA): ventricular arrhythmias
Metabolism:
Dealkylated metabolite (hepatic); less anticholinergic, less antiarrhythmic effect compared apparent compound
50% excreted unchanged, renal
Electrophysiological effects similar to quinidine gluconate (Quinaglute, Quinalan)
Similar to quinidine gluconate (Quinaglute, Quinalan) in effective ventricular and atrial tachyarrhythmia suppression
Prescribed to maintain normal sinus rhythm in patients prone to atrial fibrillation and flutter and is also used to prevent ventricular fibrillation or tachycardia.
![]() Side Effects/Toxicity
Side Effects/Toxicity
Adverse side-effect profile: different from qunidine's in that disopyramide (Norpace) is not an alpha-adrenergic receptor blocker but is anti-vagal.
Most common side effects: (anticholinergic)
Dry mouth
Urinary hesitancy
Other side effects: blurred vision, nausea
Cardiovascular:
QT interval prolongation (ECG)
Paradoxical ventricular tachycardia (quinidine-like)
Negative inotropism (significant myocardial depressive effects)--undesirable with preexisting left ventricular dysfunction (may promote congestive heart failure, even in patients with no prior evidence of myocardial dysfunction)
Disopyramide is not a first-line antiarrhythmic agent because of its negative inotropic effects
If used, great caution must be exercised in patients with congestive heart failure
Can cause torsades de pointes, a ventricular arrhythmia
Hondeghem, L.M. and Roden, D.M., "Agents Used in Cardiac Arrhythmias", in Basic and Clinical Pharmacology, Katzung, B.G., editor, Appleton and Lange, 1998, pp 216-241;Stoelting, R.K., "Cardiac Antidysrhythmic Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, 331-343
![]() Type Ib
Type Ib
Class Ib agents are often effective in treating ventricular arrhythmias. Example: lidocaine. Type Ib agents exhibit rapid association and dissociation from the channel.
![]() Mexiletine (Mexitil) (Class IB,
Sodium Channel Blocker)
Mexiletine (Mexitil) (Class IB,
Sodium Channel Blocker)
Overview
Amine analog of lidocaine (Xylocaine), but with reduced first-pass metabolism.
Suitable for oral administration
Similar electrophysiologically to lidocaine
Clinical Use:
Chronic suppression of ventricular tachyarrhythmias
Combination with a beta adrenergic receptor blocker or another antiarrhythmic drug (e.g. quinidine gluconate (Quinaglute, Quinalan) or procainamide (Procan SR, Pronestyl-SR)): synergistic effects allow:
Reduced mexiletine dosage
Decreased side effect incidence
Possibly effective: decreasing neuropathic pain when alternative medications have proven ineffective-- applications (on-label use):
Diabetic neuropathy
Nerve injury
Side effects:
Epigastric burning: usually relieved by a taking drug with food
Nausea (common)
Neurologic side effects:
Diplopia, vertigo, slurred speech (occasionally), tremor
![]() Lidocaine (Xylocaine) (Class Ib, Sodium
Channel Blocker)
Lidocaine (Xylocaine) (Class Ib, Sodium
Channel Blocker)
Overview/Pharmacokinetics:
Local anesthetic administered by i.v. for therapy of ventricular arrhythmias
Extensive first-pass effect requires IV administration
Half-life: two hours
Infusion rate: should be adjusted based on lidocaine plasma levels
Factors influencing loading and maintenance doses:
Congestive heart failure (decreasing volume of distribution and total body clearance)
Liver disease: plasma clearance -- reduced; volume of distribution -- increased; elimination half-life substantially increased (3 X or more)
Drugs that decrease liver blood flow (e.g. cimetadine, propranolol), decreased lidocaine clearance (increased possible toxicity)
Metabolism
Hepatic: some active metabolites
Cardiovascular Effects:
Site of Action: Sodium Channels
Blocks activated and inactivated sodium channels (quinidine blocks sodium channels only in the activated state)
During diastole, in normal tissue, as membrane potential returns to normal resting levels (-90 mV) lidocaine rapidly dissociates from the channel (low affinity for the channel resting state)
During diastole, in ischemic tissue, the membrane potential does not return to normal resting levels but remains partially depolarized and lidocaine remains bound (higher affinity, longer time constant for unblocking that at less negative resting potentials)
Therefore, lidocaine is more effective in suppressing activity in depolarized, arrhythmogenic cardiac tissue but has little effect on normal cardiac tissue -- the basis for this drug's selectivity.
Very effective antiarrhythmic agent for arrhythmia suppression associated with depolarization (e.g., digitalis toxicity or ischemia)
Comparatively ineffective in treating arrhythmias occurring in normally polarized issue (e.g., atrial fibrillation or atrial flutter)
No significant effect on QRS or QT interval or on AV conduction (normal doses)
Lidocaine (Xylocaine) decreases automaticity by reducing the phase 4 slope and by increasing threshold.
Clinical Uses:
Suppression of ventricular arrhythmias (limited effect on supraventricular tachyarrhythmias)
Suppression of reentry-type rhythm disorders:
Premature ventricular contractions (PVCs)
Ventricular tachycardia
May reduce incidence of ventricular fibrillation during the initial time frame following acute myocardial infarction; no evidence to support prophylactic use and myocardial infarction
![]() Side Effect/Toxicities
Side Effect/Toxicities
Overdosage:
Vasodilation
Direct cardiac depression
Decreased cardiac conduction -- bradycardia; prolonged PR interval; widening QRS on ECG
Major side effect -- neurological
Large doses, rapidly administered can result in seizure.
Factors that reduce seizure threshold for lidocaine:
Hypoxemia, hyperkalemia, acidosis
Otherwise: CNS depression, apnea.
![]() Tocainide (Class I, Sodium Channel
Blocker)
Tocainide (Class I, Sodium Channel
Blocker)
Amine analog of lidocaine, similar to mexiletine, orally active --but with reduced first-pass metabolism.
Used for chronic suppression of ventricular tachyarrhythmias
Electrophysiologically similar to lidocaine
Similar to mexiletine: tocainide + if beta-adrenergic receptor blocker or another antiarrhythmic drug: synergism
e.g. Combination with quinidine may increase efficacy and diminish adverse effects.
![]() Side Effects:
Side Effects:
Profile similar to mexiletine
Suitable for oral administration, but RARELY USED due to possibly fatal bone marrow aplasia and pulmonary fibrosis.
Tremor and nausea are major dose-related adverse side effects
Excreted by the kidney, accordingly dose should be reduced in patients with renal disease
Stoelting, R.K., "Cardiac Antidysrhythmic Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, 331-343
Phenytoin
Overview
Effective in suppression of ventricular arrhythmias associated with digitalis toxicity
Less effective than quinidine, procainamide, or lidocaine, in treatment of ventricular arrhythmias due to other etiologies
Pharmacokinetics:
Routes of administration: oral, or IV
Normal saline preferred -- phenytoin may precipitate in 5% dextrose in water
Slow IV injection into large peripheral or central vein preferable-- decreased chance of:
Discomfort
Thrombosis at injection site
Hepatic Metabolism --hydroxylation and conjugation (glucuronidation):
Elimination half-life: approximately 24 hours
Impaired hepatic function may cause excessive phenytoin blood levels
Mechanism of Action/Cardiac Effects:
Electrophysiological effects on automaticity and conduction velocity--somewhat like lidocaine
Shortens QT interval more than any other antiarrhythmic agent
No significant effect on ST-T waves or QRS complex
No significant myocardial depression
Improvement in AV Node Conduction;
Depression of SA Nodal Activity
Drug-Drug Interaction:
Significant SA nodal depression may occur when combining those volatile anesthetics that depress SA nodal activity and phenytoin
Drugs that lower phenytoin levels:
Barbiturates (mechanism:metabolizing enzyme induction)
Drugs that increase phenytoin level (inhibit metabolism):
Warfarin, phenylbutazone, isoniazid
Side effect/Toxicities:
Primary Toxicity: CNS disturbance (particularly cerebellar-- dose correlated > 18 ug/ml-- exceeding this concentration is unlikely to improve cardiac rhythm)
CNS symptoms:
Ataxia, vertigo, slurred speech, sedation, nystagmus, confusion
Partial inhibition of insulin secretion: enhances blood glucose levels in hyperglycemic patients
Stoelting, R.K., "Cardiac Antidysrhythmic Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, 331-343
![]() Type Ic: Type Ic drugs slowly dissociate
from resting sodium channels
Type Ic: Type Ic drugs slowly dissociate
from resting sodium channels
Flecainide (Tambocor)--( Na+ and K+ Channel Blocker)
Overview:
Fluorinated local anesthetic analog of procainamide (Procan SR, Pronestyl-SR)
More effective than quinidine gluconate (Quinaglute, Quinalan) or disopyramide (Norpace) in:
Suppressing ventricular tachycardia
Suppressing ventricular premature contractions
Pharmacokinetics:
Oral absorption: excellent
Long elimination half-time (approximately 20 hours)
25% flecainide: excreted unchanged (kidneys)
Hepatic metabolism: weakly active metabolites
Factors reducing flecainide elimination:
Congestive heart failure
Renal failure
Cardiac Effects/Clinical Use:
Suppression ventricular tachycardia and ventricular premature contractions
Effective in management of atrial tachyarrhythmias
Effective in tachyarrhythmias associated with Wolff-Parkinson-White syndrome (suppression of conduction bypass tracts)
Chronic flecainide (Tambocor) treatment following myocardial infarction not recommended:
Increased incidence of sudden death in treated patients
In CAST, flecainide increased mortality in patients recovering from myocardial infarction.
Flecainide: should be reserved for management of life-threatening arrhythmias
Slight/moderate negative inotropic property
Proarrhythmic effects in patients with preexisting left ventricular function deficiency
Electrophysiology:
Prolongation of PR interval (ECG)
Prolongation of QRS complex (> 25%)
Sinoatrial nodal depression (similar to beta-adrenergic blockers and calcium channel blockers)
Most common:vertigo and difficulty in visual accommodation
Most serious of adverse effects is induction of potentially lethal arrhythmias such as reentrant ventricular tachyarrhythmias.
Stoelting, R.K., "Cardiac Antidysrhythmic Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, 331-343
Overview:
A benzofurane derivative, 37% iodine by weight, structurally similar to thyroxine
May cause hypothyroidism or hyperthyroidism (frequency: 2%-4%)
Insidious development
Patients with previous thyroid dysfunction: more likely to develop amiodarone-mediated thyroid effects
Hyperthyroidism: most readily evidenced by increased plasma level of triiodothyronine
Secondary to iodine release from parent drugs;
Often refractory to conventional treatment
Intolerant of beta-adrenergic receptor blockade (because of underlying cardiac disease)
Following failed medical management: surgical thyroidectomy is appropriate
Bilateral superficial cervical plexus block has been used for anesthetic management of subtotal thyroidectomy in this patient group
Hypothyroidism: most readily evidenced by increased plasma level of thyroid-stimulating hormone (TSH)
May interfere with certain radiologic procedures (Iodine accumulation)
Approved for use only in treatment of serious ventricular arrhythmias (USA)
Also used for refractory supraventricular arrhythmias
Numerous adverse effects.
Metabolism and Excretion
Long elimination halftime: 29 days
Minimal renal excretion
Principal metabolite (desmethylamiodarone) -- longer elimination halftime compared to amiodarone
Extensive protein binding
Amiodarone concentrated in the myocardium (10-50 times plasma concentration)
Cardiovascular Properties and Uses:
Used in patients with ventricular tachycardia or fibrillation resistant to treatment with other drugs.
Effective inhibitor of abnormal automaticity.
Oral administration, preoperatively, reduces likelihood of atrial fibrillation following cardiac surgery.
Suppresses tachyarrhythmias associate with Wolff-Parkinson-White syndrome
secondary to depression of conduction in the AV node and accessory bypass tracts.
Similar to β-blockers (unlike most class I antiarrhythmics), amiodarone decreases mortality after myocardial infarction
Antiarrhythmic effectiveness begins within 72 hours following initiation of oral treatment; nearly immediate effect following IV administration
Following discontinuation of chronic oral therapy: pharmacological effects may last up to two months (long elimination half-time)
Mechanism of Action
Blocks sodium and potassium channels and prolongs action potential duration.
Prolongs effective refractory period in:
SA node
AV node
Ventricle
Atrium
His-Purkinje system
Accessory bypass tracts (Wolff-Parkinson-White syndrome)
Vascular Effects
Noncompetitive alpha and beta adrenergic receptor blocker
Systemic vasodilation
Antianginal properties, secondary to coronary vasodilation
Pulmonary:
Most serious adverse effect seen in long-term therapy is a rapidly progressive pulmonary fibrosis which may be fatal
Frequency: 5%-15% treated patients
Mortality rate: 5% to 10%
Cause: unknown (possibly related to amiodarone-mediated generation of free oxygen radicals in the lung)
Two types of amiodarone-pulmonary toxicity clinical presentations:
More common: Slow, insidious, progressive dyspnea, cough, weight loss, pulmonary infiltration (chest x-ray)
Acute onset: dyspnea, cough, arterial hypoxemia.
Anesthetic Implications: pulmonary
Suggested restriction of inspired oxygen concentration in patients receiving amiodarone and undergoing general anesthesia close level possible while retaining adequate systemic oxygenation
Postoperative pulmonary edema has been reported in patients treated with amiodarone chronically-- resembles acute onset form of amiodarone toxicity.
In patients with preexisting amiodarone-cause pulmonary damage are at increased risk for adult respiratory distress syndrome following surgery requiring cardiopulmonary bypass.
Cardiovascular Effects:
Prolongation of QT interval (ECG); increased incidence of ventricular tachyarrhythmias (including torsades de pointes)
Bradycardia (atropine-resistant)
Catecholamine responsiveness: diminished due to alpha and beta-receptor blocking activity
Hypotension; A-V block (following IV administration)
Anesthetic Implications: cardiovascular
With general anesthesia -- enhanced antiadrenergic action, presentation as:
A-V block, sinus arrest, decrease cardiac output, hypotension
Sinus arrest more likely in the presence of anesthetics that inhibit SA nodal automaticity (e.g. lidocaine, halothane)
Consideration should be given for temporary ventricular pacemaker and sympathomimetic administration (e.g. isoproterenol) for patients taking amiodarone and scheduled undergo surgery.
Ocular and other Side Effects:
Corneal microdeposits: common;usually no visual impairment
Photosensitivity, rash: 10% frequency
Rare: cyanotic discoloration (slate-gray facial pigmentation)
Neurological:
Peripheral neuropathy; sleep disturbance, headache, tremor, some skeletal muscle weakness
Drug-drug interaction
Potent inhibitor of hepatic metabolism or renal elimination of many drugs.
Warfarin, quinidine gluconate (Quinaglute, Quinalan), procainamide (Procan SR, Pronestyl-SR) and digoxin (Lanoxin, Lanoxicaps) are examples of drugs which may require dosage reduction during amiodarone (Cordarone).
Amiodarone (Cordarone) displaces digoxin (Lanoxin, Lanoxicaps) from protein binding sites
Digoxin (Lanoxin, Lanoxicaps) levels may increase as much as 70%
Digoxin (Lanoxin, Lanoxicaps) dose should be decreased as much as 50% when amiodarone is administered concurrently
Hondeghem, L.M. and Roden, D.M., "Agents Used in Cardiac Arrhythmias", in Basic and Clinical Pharmacology, Katzung, B.G., editor, Appleton and Lange, 1998, pp 216-241; Stoelting, R.K., "Cardiac Antidysrhythmic Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, 331-343
|
|
|
|
|
|
|