Medical Pharmacology Chapter 2 General Principles: Adverse Effects
|
|
|
|
|
|
|
|
|
-
Audio Overview: Drug-Drug Interactions and Adverse Effects (Extended)
-
Audio Overview: Drug-Drug Interactions and Adverse Effects (Brief)
Why Adverse Effects Are Pharmacology, Not Bad Luck
Adverse drug reactions (ADRs) are not random misfortunes.
Most adverse effects are predictable consequences of pharmacological mechanism.
The same receptor interactions, enzyme inhibitions, and signal transduction effects that produce therapeutic benefit also produce harm when they occur at unintended sites, at excessive intensity, or in vulnerable patients.
A prescriber who understands pharmacology can anticipate, recognize, and often prevent ADRs rather than simply react to them.
This section establishes the classification and mechanisms of Adverse Drug Reactions (ADRs), examines the clinical consequences of pharmacokinetic drug–drug interactions, particularly through the Cytochrome P450 Drug Metabolizing System (CYP enzyme system), and identifies the drug classes and patient populations at highest risk.
The overview presented describe the basis for rational prescribing and thus is applicable in the clinical setting.1
Classification of Adverse Drug Reactions: The A–D System
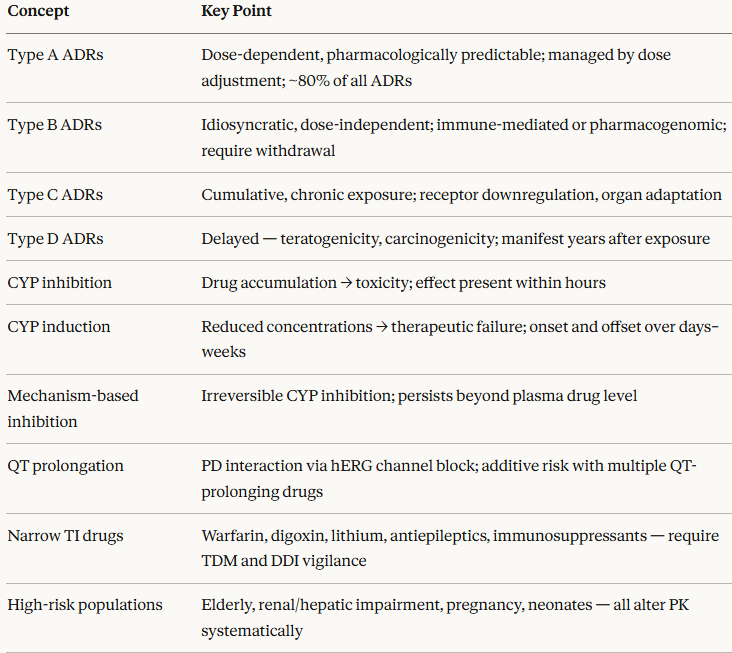
The most widely used classification system divides ADRs into four types based on their mechanism and predictability.1,2
Type A represent Augmented (Pharmacological) Reactions
Type A reactions are dose-dependent, predictable from the drug's known pharmacology, and account for approximately 80% of all ADRs.
They represent an exaggeration of the drug's intended pharmacodynamic effect, either at the target organ or at secondary sites.
Because they are mechanistically predictable, they can be anticipated and managed by dose adjustment.
Examples
Hypoglycemia from insulin or sulfonylureas (exaggerated glucose-lowering effect)
Bradycardia and heart block from β-blockers (exaggerated negative chronotropic effect)
Bleeding from anticoagulants (exaggerated anticoagulant effect)
Hypotension from antihypertensives; respiratory depression from opioids.
The clinical implication: for Type A reactions, dose reduction, route change, or addition of a pharmacological antidote (such as the opioid antagonist naloxone (Narcan) for opioid-induced respiratory depression) is usually effective.
Understanding the receptor mechanism immediately points toward the corrective strategy.
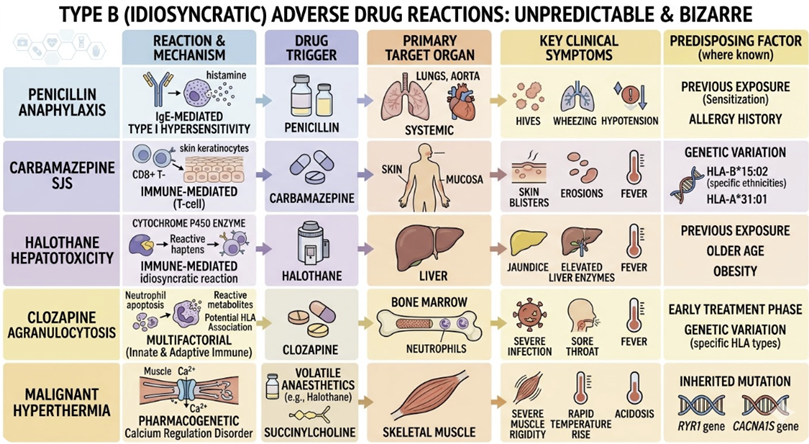
Type B refer to Bizarre (Idiosyncratic) Reactions
Type B reactions are dose-independent, unpredictable from the drug's pharmacology, and occur only in susceptible individuals.
They are less common than Type A reactions but are often more severe, with higher mortality.
The mechanisms include immune-mediated hypersensitivity and pharmacogenomic susceptibility.
Examples:
Penicillin anaphylaxis (IgE-mediated type I hypersensitivity)
Carbamazepine-induced Stevens-Johnson syndrome (immune-mediated)
Halothane-induced hepatotoxicity
Clozapine-induced agranulocytosis
Malignant hyperthermia triggered by volatile anaesthetics or suxamethonium (a rare pharmacogenomic reaction involving ryanodine receptor mutations).
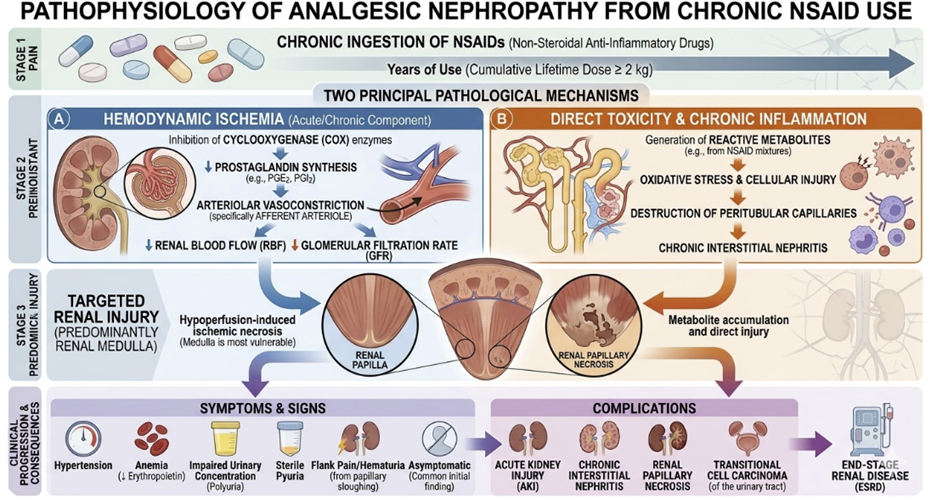
Type C refers to Chronic (Cumulative) Reactions
Type C reactions occur with long-term drug use and are related to cumulative dose and duration of exposure rather than any single dose.
They reflect the consequences of prolonged pharmacodynamic receptor engagement such as downregulation, organ adaptation, or structural tissue changes.
Examples:
Adrenal suppression from chronic systemic glucocorticoids (HPA axis downregulation from prolonged nuclear receptor activation)
Osteoporosis from long-term corticosteroids
Tardive dyskinesia from years of antipsychotic use (dopamine receptor supersensitivity from prolonged D2 blockade)
Analgesic nephropathy from chronic NSAID use.
The clinical implication: Type C reactions require long-term monitoring strategies and, where possible, planned dose tapering rather than abrupt discontinuation to allow receptor upregulation to normalize.
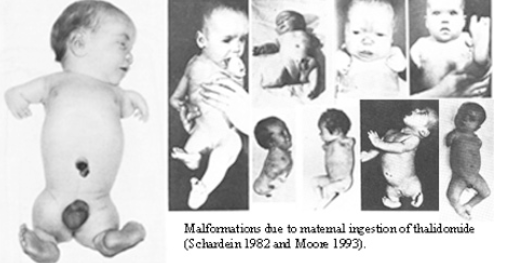
Type D refers to Delayed Reactions
Type D reactions manifest long after drug exposure, sometimes years to decades.
They include teratogenicity (structural defects from drug exposure during organogenesis), carcinogenicity (drug-induced malignancy), and mutagenicity.
The canonical historical example is thalidomide, prescribed as a sedative in the late 1950s, withdrawn in 1961 after causing limb reduction defects (phocomelia) in thousands of children whose mothers had taken the drug during the first trimester.
Attribution
Public domain, via Wikimedia Commons
https://commons.wikimedia.org/wiki/File:Thalidomide_effects.jpg
Follow page link above for study author
Thalidomide's teratogenicity was not detectable by the pharmacological testing methods of the era, and its discovery fundamentally transformed drug regulatory requirements worldwide.
Tamoxifen, used to prevent and treat estrogen receptor-positive breast cancer, carries a small but real risk of endometrial carcinoma from its partial estrogen agonist activity in uterine tissue, a Type D reaction requiring surveillance1,2
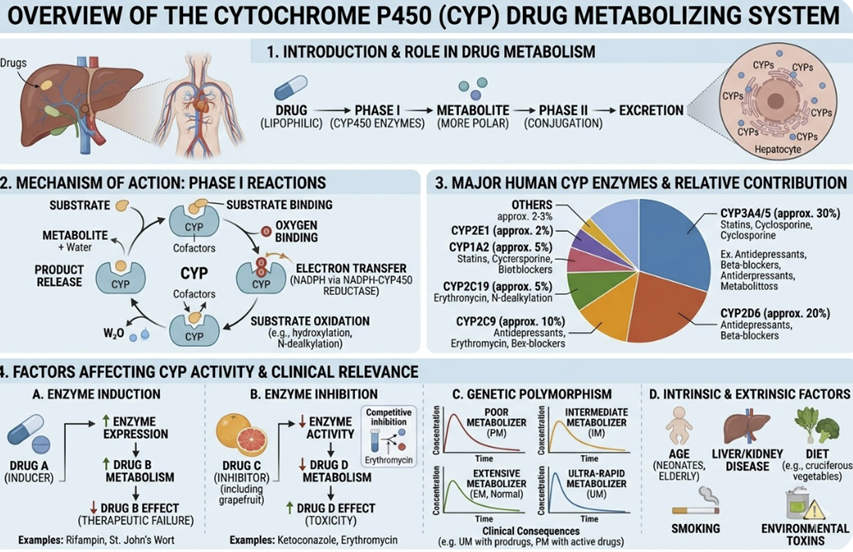
Pharmacokinetic Drug–Drug Interactions: The CYP System
-
Drug–drug interactions (DDIs) at the pharmacokinetic level occur when one drug alters the absorption, distribution, metabolism, or excretion of another, changing its plasma concentration and therefore its pharmacodynamic effect.
-
The most
clinically important mechanism is metabolic interaction through the
cytochrome P450 (CYP) enzyme system.1,2,3
-
-
The hepatic CYP enzyme system comprises a family of membrane-bound heme-containing oxidases responsible for the Phase I metabolism of approximately 70–80% of clinically used drugs.
-
The five most important isoforms in clinical pharmacology are CYP3A4, CYP2D6, CYP2C9, CYP2C19, and CYP1A2.
-
-
-
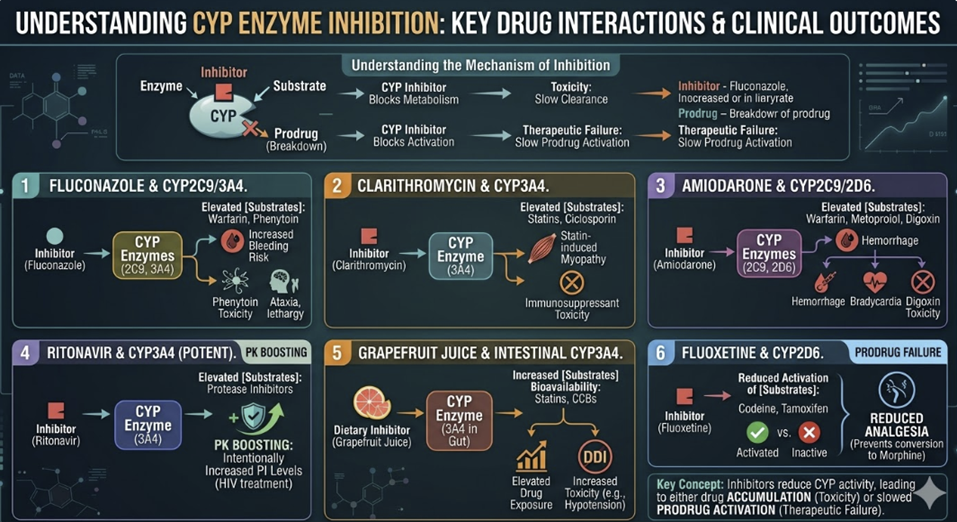
CYP inhibition occurs when one drug reduces the metabolic activity of a CYP isoform, impairing the metabolism of co-administered drugs (substrates) processed by the same enzyme.
-
The result is drug accumulation reflected in elevated plasma drug
levels that may produce toxicity at standard doses.
-
-
Inhibition can be:
-
Competitive (the inhibitor competes with the substrate for the enzyme's active site, reversibly)
-
Mechanism-based (the inhibitor is metabolized to a reactive intermediate that irreversibly inactivates the enzyme (also called suicide inhibition), or
-
Mixed.
-
-
Mechanism-based inhibitors are particularly important because their effect persists beyond their plasma half-life, requiring new enzyme synthesis for recovery.
-
Inhibition can be:
-
Competitive (the inhibitor competes with the substrate for the enzyme's active site, reversibly)
-
Mechanism-based (the inhibitor is metabolized to a reactive intermediate that irreversibly inactivates the enzyme — also called suicide inhibition), or
-
Mixed.
-
Mechanism-based inhibitors are particularly important because their effect persists beyond their plasma half-life, requiring new enzyme synthesis for recovery.
-
-
-
-
Clinically important examples of CYP inhibition:3,4
-
-
-
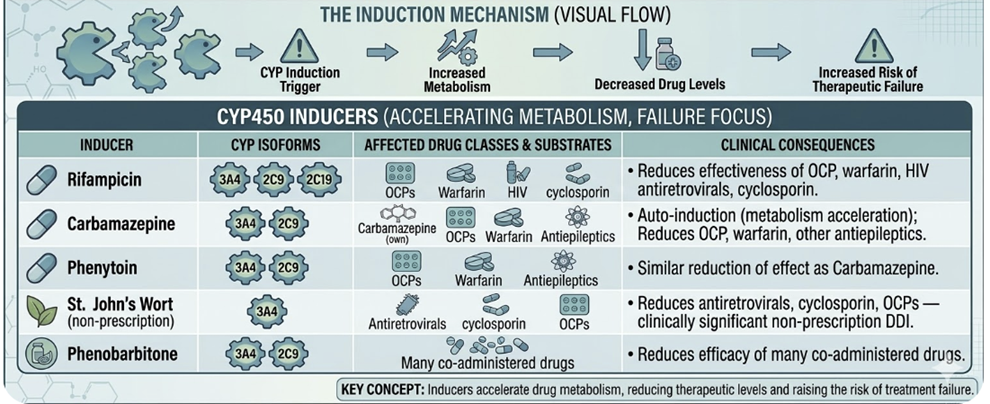
CYP induction occurs when a drug up-regulates the transcription of CYP genes, increasing enzyme expression and accelerating the metabolism of co-administered substrates.
-
The result is reduced plasma drug concentrations which may prevent an adequate therapeutic effect.
-
-
Induction is always a delayed phenomenon since new protein synthesis is required. New protein synthesis may take days to weeks to reach maximum effect.
-
Similarly, when the inducer is discontinued, enzyme levels return to baseline over days to weeks.
-
-
Clinically important CYP inducers:3,4
-
-
The most
dangerous induction interactions are those where therapeutic failure
cannot be detected clinically until a very serious medical event
occurs.
-
An example: Rifampicin reducing cyclosporin concentrations to subtherapeutic levels in a transplant patient, precipitating acute rejection.
-
-
-
Pharmacodynamic Drug–Drug Interactions
-
Not all clinically important drug interactions are pharmacokinetic.
-
Pharmacodynamic interactions occur when two drugs act on the same physiological system, producing effects that are greater or smaller than either drug alone, without any change in plasma concentrations.1,2
-
Synergism (Additive or Supra-additive)
-
Additive interactions occur when two drugs produce combined effects equal to the sum of their individual effects.
-
Supra-additive (synergistic) interactions produce combined effects greater than the arithmetic sum — one drug amplifies the effect of another.
-
-
Examples
-
Opioids combined with benzodiazepines produce supra-additive CNS and respiratory depression.
-
This combination can result in opioid overdose deaths with combination more dangerous than dose-equivalent opioids alone.
-
-
Beta-lactam antibiotics combined with aminoglycosides produce synergistic bactericidal activity against certain organisms by targeting bacterial cell wall synthesis (beta-lactam) and protein synthesis (aminoglycoside) simultaneously.
-
-
-
-
Pharmacodynamic antagonism occurs when two drugs produce opposing effects reducing each other's activity.
-
This effect can be therapeutically exploited (for example naloxone antagonizes opioid toxicity) or clinically harmful (for example non-steroidal anti-inflammatory drugs reduce the ACE inhibitor mediated antihypertensive effects through prostaglandin-mediated reduction of renal sodium excretion).
-
-
-
QT Prolongation and Torsades de Pointes
-
One of the most clinically important pharmacodynamic interactions is QT prolongation resulting from combined effect of multiple drugs that block cardiac hERG (human Ether-a-go-go Related Gene) potassium channels, thus delaying ventricular repolarisation.
-
Individual drugs at therapeutic doses may produce modest QT prolongation but in combination may produce pathological prolongation as well as potentially fatal arrhythmia torsades de pointes.
-
Prolonged QT Interval; Torsades de Pointes (TdP) 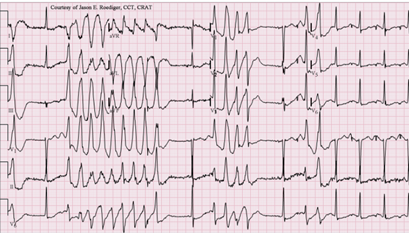
-
Attribution
-
Jer5150, CC BY-SA 3.0 <https://creativecommons.org/licenses/by-sa/3.0>, via Wikimedia Commons June 3, 2012
-
https://commons.wikimedia.org/wiki/File:Torsades_de_Pointes_TdP.png
-
-
-
-
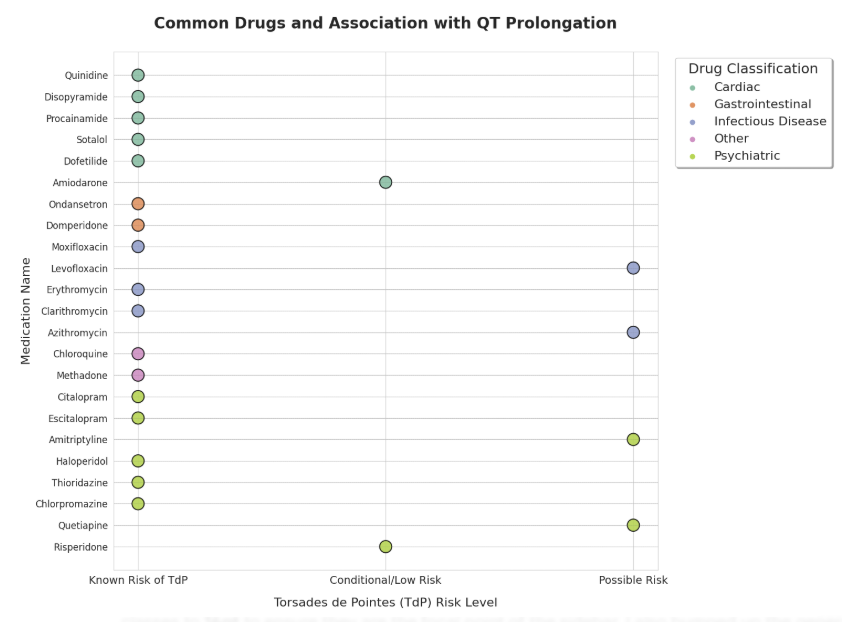
Classes of drugs implicated in QT prolongation include:
-
Antipsychotics (haloperidol, quetiapine)
-
Antiarrhythmics (amiodarone, sotalol, quinidine)
-
Antibiotics (azithromycin, fluoroquinolones)
-
Antifungals (fluconazole)
-
Antiemetics (domperidone, ondansetron), and
-
Antidepressants (TCAs, citalopram at higher doses).
-
-
Prescribers should check for QT-prolonging drug combinations, particularly in patients with baseline QT prolongation, electrolyte abnormalities (hypokalaemia, hypomagnesemia), structural heart disease, or bradycardia.5
-
-
-
-
High-Risk Drug Combinations and Narrow Therapeutic Index Drugs
-
Certain drugs require particularly careful monitoring and interaction screening because of their narrow therapeutic index which represents a small margin between therapeutic and toxic plasma concentrations.1,2
-
Anticoagulants (warfarin, direct oral anticoagulants)
-
Warfarin is a CYP2C9 substrate with a narrow therapeutic index and an enormous number of pharmacokinetic and pharmacodynamic interactions.
-
Any change in
CYP2C9 inhibitors, inducers, dietary vitamin K intake, or
co-administration of antiplatelet agents requires INR reassessment.
-
Direct Oral Anticoagulants (DOACs) have fewer pharmacokinetic interactions but are substrates of P-glycoprotein and CYP3A4; interactions with rifampicin, antifungals, and certain antiepileptics are clinically significant.
-
Examples of DOACs include apixaban (Eliquis), rivaroxaban (Xarelto) and dabigatran (Pradaxa)
-
-
-
-
Immunosuppressants (ciclosporin, tacrolimus, sirolimus)
-
CYP3A4 substrates with narrow therapeutic indices:
-
Sub-therapeutic levels risk organ rejection
-
Supratherapeutic levels cause nephrotoxicity, neurotoxicity, and infection.
-
-
The CYP3A4 interactions above (particularly rifampicin, clarithromycin, and azole antifungals) require immediate dose adjustment and enhanced monitoring.
-
-
Digoxin
-
Digoxin is a narrow therapeutic index cardiac glycoside whose toxicity (arrhythmias, visual disturbance, nausea) occurs at plasma concentrations only marginally above the therapeutic range.
-
Amiodarone and clarithromycin both increase digoxin concentrations by inhibiting renal P-glycoprotein-mediated tubular secretion, a transporter rather than CYP-mediated interaction.
-
-
Antiepileptics
-
Many antiepileptics are both CYP inducers which may decrease concentrations of co-prescribed drugs and CYP substrates.
-
Phenytoin, carbamazepine, and valproate have complex, bidirectional DDI profiles and narrow therapeutic indices.
-
Combination antiepileptic therapy requires Therapeutic Drug Monitoring (TDM).
-
-
-
-
Lithium
-
Renally excreted unchanged
-
Accumulates rapidly with any reduction in renal blood flow or tubular function
-
NSAIDs (which reduce renal prostaglandin synthesis), ACE inhibitors, and diuretics all raise lithium concentrations.
-
Dehydration, a new disease/infection developing while a patient is already suffering from another, often chronic condition (intercurrent illness), and dietary sodium restriction are common precipitants of lithium toxicity in therapeutically managed patients.
-
-
Patient Populations at Elevated ADR Risk
-
Some patient populations have altered pharmacokinetics or pharmacodynamics that systematically increase their Adverse Drug Reaction (ADR) risk.1,2,6
-
Elderly patients
-
Age-related reduction in hepatic CYP450 activity (approximately 30–40% reduction), reduced renal GFR, decreased plasma albumin (increasing free fractions of protein-bound drugs), and reduced body water and lean mass (altering volume of distribution) collectively narrow the margin between therapeutic and toxic drug levels.
-
Polypharmacy (patient prescribed many drugs and especially prevalent in older adults) multiplies Drug-Drug Interaction (DDI) risk.
-
The START/STOPP criteria provide evidence-based guidance for prescribing optimization in elderly patients.
-
-
-
Patients with renal impairment
-
Reduced renal clearance increases concentrations of renally excreted drugs (aminoglycosides, digoxin, lithium, metformin, many antibiotics, direct oral anticoagulants).
-
Dose reduction or interval extension is required, calculated against the patient's estimated GFR.
-
-
-
Patients with hepatic impairment
-
Reduced hepatic CYP450 activity, reduced albumin synthesis which may increase free drug fractions, and portal-systemic shunting associated with increased bioavailability of high-extraction drugs, all alter drug metabolism.
-
The Child-Pugh score provides a clinical framework for dose adjustment in hepatic impairment.7
-
-
-
Pregnant patients
-
Expanded plasma volume, accelerated renal clearance, altered CYP expression, and the teratogenic risk of many drugs require individualized prescribing with reference to current teratogenicity data.
-
The
thalidomide disaster established the principle that placental
transfer of drugs must be assumed unless proven otherwise.
-
-
-
Neonates and infants
-
Incompletely developed hepatic enzyme systems (particularly UGT enzymes and CYP3A4) and reduced renal function substantially impair drug clearance.
-
Drug
doses cannot be simply scaled from adult doses without
accounting for these maturational pharmacokinetic
differences.
-
-
-
 |
|
|
|
|
|
|
References
|
|