General Principles: Overview and Introduction
|
|
|
|
|
|
|
|
|
-
The Scope of Medical Pharmacology
-
Medical pharmacology focuses on the use of particular chemicals, drugs, used both for prevention and treatment of disease.9a
-
New drugs are identified or developed for a number of different reasons including new drug targets identified as molecules central to pathological processes.
-
As the molecular basis of pathologies become appreciated in greater detail, new potential targets for drugs are being identified.
-
The discovery of new diseases also promote the need for new treatment approaches which sometimes involve new drugs.
-
Also, the development of resistance to standard of care drugs by bacteria, parasites or even by cancer cells necessitate new drug therapies.9a
-
-
-
More recently, some new drugs have taken the form of antibodies which may inactivate protein targets central to disease and disease progression.
-
Other new drugs, perhaps inhibitors of various kinases, continue to be best described as "small molecules."
-
-
-
-
-
Drugs interact with receptors, usually proteins, and as a consequence the receptors are either activated or blocked from activation.
-
Drug-Receptor Interactions 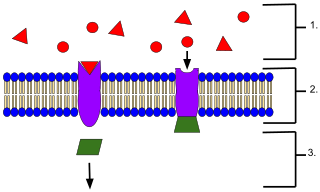
-
"An example of membrane receptors.
-
Ligands located outside the cell.
-
Ligands connect to specific receptor proteins based on the shape of the active site of the protein.
-
The receptor releases a messenger once the ligand has connected to the receptor."
-
-
Attribution: Wyatt Pyzynski, CC BY-SA 4.0 <https://creativecommons.org/licenses/by-sa/4.0>, via Wikimedia Commons
-
https://commons.wikimedia.org/wiki/File:Receptor_(Biochemistry).svg
-
-
A few drugs may be described as "chemical antagonists" given that they interact directly with other drugs.7
-
Another drug category is that of "osmotic substances," interacting with water molecules.
-
-
Sometimes drug is synthesized by the body, naturally occurring.
-
Hormones represent such a class.
-
-
Many drugs are chemicals which are not made by the body and as such are described as xenobiotics.7
-
-
-
There are several classes (protein) of human receptors.3,5
-
These include:
-
Endocrine and paracrine factor receptors
-
Voltage-gated ion channels
-
Membrane transporters
-
-
Most drugs produce effects by interacting with receptors and enzymes.
-
 Another
broad class of receptors include:
Another
broad class of receptors include:-
Structural proteins
-
Enzyme-interacting proteins
-
Transcription factors
-
Ribosomes, DNA and RNA
-
Some examples:
-
Drugs that interact with enzymes: lisinopril (as well as other angiotensin-converting enzyme (ACE) inhibitors).
-
Drugs that inhibit HMG-CoA reductase (3-hydroxy-3-methyl-glutaryl-CoA reductase) including atorvastatin and lovastatin.
-
Drugs that interact with voltage-gated ion channels (and other ion channels) include: amlodipine and gabapentin, which inhibit neural voltage-gated Ca2+ channels) and glipizide (enhancing ATP-regulated K+ channels).3,5
-
-
Drugs that activate receptors are typically termed "agonists" whereas those drugs that block the receptors are termed "antagonists." 2
-
Drug-receptor binding is usually a readily reversible process.
-
However, certain drugs interact covalently with their receptor thus inactivating it.2
-
In this circumstance, a new receptor molecule must be synthesized, replacing the inactivated protein.
-
An example of a covalent drug-receptor interaction is that of aspirin and its acetylation of cyclooxygenases.
-
Aspirin
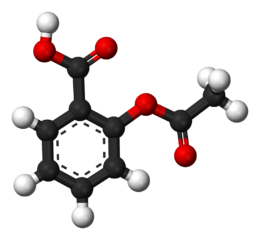
-
Benjah-bmm27, Public domain, via Wikimedia Commons
-
https://commons.wikimedia.org/wiki/File:Aspirin-3D-balls.png
-
-
Irreversible acetylation of cyclooxygenases diminishes prostaglandin production accounting for aspirin's anti-inflammatory activity as well as decreasing thromboxane synthesis which accounts for the antiplatelet activity of aspirin.2
-
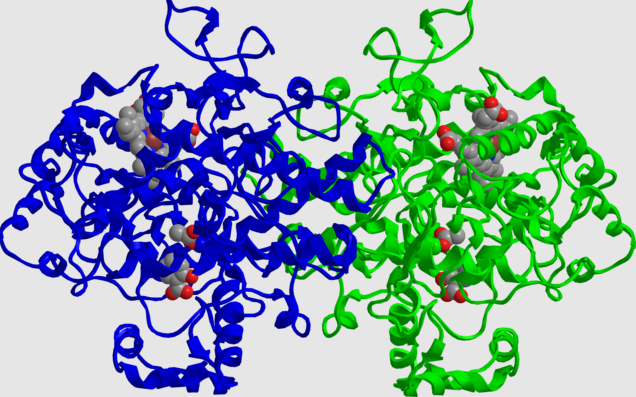
-
"Structure COX2 (also called prostaglandin H synthase) inactivated by aspirin. The molecule is a dimer, the blue and green halves are identical. In each monomer, the active site serine has been acetylated, inactivating it. Also visible in each monomer is the salicylic acid from which the acyl group came, and the heme cofactor."
-
Attribution: Jeff Dahl, Public domain, via Wikimedia Commons.
-
COX1 and COX2 represent 2 distinct cyclooxygenase isoenzymes.
-
Cyclooxygenase catalyzes the conversion to arachidonic acid to inflammatory prostaglandins (PGs).
-
Aspirin is nonselective and inhibits both isoforms.
-
Aspirin acetylates a serine residue of cyclooxygenase-1 (COX1) which blocks the formation of thromboxane A2 in platelets, thus decreasing platelet aggregation.
-
This effect of aspirin accounts for a reduction in the likelihood of cerebrovascular and coronary artery thrombosis.8
-
-
-
Aspirin is less effective in competitively inhibiting cyclooxygenase-2 (COX2).
-
Inhibition of COX2 is responsible for the anti-inflammatory effects of the nonsteroidal anti-inflammatory agents (NSAIDs).
-
Examples of other drugs that exhibit greater selectivity in COX2 inhibition include celecoxib (Celebrex) and rofecoxib (Vioxx).9
-
-
-
-
-
-
-
-
-
Non-Covalent Drug-Receptor Interactions
-
The
tendency of drugs to bind to receptors describe affinity.
-
Given that the receptors are typically proteins, only certain regions of the receptor protein defines the drug binding domain or binding site.
-
Factors that influence binding are varied, including the ionization state of amino acids associated with the binding site (PKa), as well as hydrophobicity and hydrophilicity of the local environment.1
-
Drug receptor binding often involves multiple bond types with differing associated bond strengths.1
-
The least strong association is due to van der Waals forces that reflect changing electron densities in the molecule.
-
Hydrogen bonding occurs when hydrogen atoms, binding to oxygen or nitrogen, become more polarized (positively) facilitating association with sulfur, nitrogen or oxygen (negatively polarized).
-
Ionic bonds occur between atoms which have an overall negative charge (electronic excess) and thus favor association with atoms exhibiting a relative electron deficiency.
-
-
-
Covalent bonding involves electron sharing between atoms.1
-
-
-
-
-
-
-
The drug molecule interacts with the receptor binding domain in a way that exhibit structural complementarity between the two.10
-
Sometimes this complementarity is referred to the relationship between a lock and its key.
-
Another important aspect is the relationship between drug action and its stereochemistry.
-
Stereoisomers refer to molecules exhibiting both the same number and types of atoms and even the same bond arrangement but show different three-dimensional spatial structures.10
-
Stereoisomers are composed of two types: enantiomers and diastereomers.
-
Enantiomers refer
to molecular pairs in which the atoms are not super-imposable i.e.
nonsuperimposable mirror images. Diastereomers consider all the other
stereoisomers that are not enantiomers.10
-
Definition: Chirality- "handedness"; (from the Greek cheir meaning the "hand"); molecules which are not superimposable on (cannot be made to coincide with) their mirror image. achiral molecules can be superimposed on its mirror image.
-
-
Chirality has to do with "right or left handedness"
-
If an item appears identical to its image in a mirror it is said to be "achiral"; an example would be a water glass.
-
The basic issues is a comparison between an object and its reflection.
-
If an object is different from its reflection it is said to be chiral.
-
For instance, ones left-hand and right hand are chiral'
-
-
-
-
Many drugs, over 50% of all drugs, are chiral (existing as enantiomeric pairs).
-
Enantiomers (molecules having opposite shapes) are pairs of molecules existing in forms that are mirror images of each other (right and left-hand) but that cannot be superimposed.
-
Other than lack of superimposition, enantiomers are chemically identical, but may be distinguished by the direction in which they rotate polarized light, either dextro (d or +) or levo (l or -).
-
The light rotational effect is determined in solution.
-
-
-
"Plane-Polarized Light Rotation:
-
Plane-polarized light is light in which all wave vibrations have been filtered out except for those in one plane.
-
Passing light through a polarized lens allows only one wave direction to be transmitted.
-
When the plane-polarized light wave is then passed through a solution containing a single enantiomer of a compound the light is rotated by a certain amount either clockwise or counter clockwise.
-
-
-
"Optical Rotation is the rotation of plane-polarized light.
-
Optically active compounds are chiral compounds which possess the ability to rotate plane-polarized light.
-
A polarimeter is an instrument which polarizes light and then shows the angle of rotation of plane-polarized light by an optically active compound.
-
Specific rotation is the degree of rotation by 1.00 gram of compound in 1.00 mL of solution measured in a tube with a 1 decimeter path length.
-
The amount of rotation is also dependent on the temperature (usually room temperature), wavelength (typically the D line of sodium, 589.3 nm) and solvent."
-
Attribution: Dr. Nanette Wachter-Jurcsak, Hoftra University
-
-
-
-
About 25% of drugs presently on the market represent some type of isomeric combination.
-
Most of these isomeric mixtures are racemates, defined as a mixture with equal amounts of both drug enantiomers present.
-
Administration
of a drug composed of differing enantiomers may display
pharmacokinetic and pharmacodynamic characteristics that reflect
contributions of the differing enantiomeric forms.
-
-
Sometimes an individual isomer may be metabolized differently, absorbed differently and exhibit differing degrees of serum protein binding compared to the other isomer.10
-
-
-
-
-
-
In some cases, racemic drugs exhibit one major pharmacologically active enantiomer.11
-
Within this group a number of cardiovascular drugs have been identified and are routinely used to treat heart failure, hypertension, as well as arrhythmias.
-
These agents include Ca2+ channel antagonists, β-adrenergic antagonists and ACE inhibitors (Angiotensin-Converting Enzyme inhibitors).11
-
Some examples:
-
In general the levorotary-isomer of β-antagonists exhibits higher potency for blockade of β-adrenergic receptors compared to the dextrorotary-form.
-
Note that the D- L designation describes the whole molecule; whereas, the R/S system describes the configuration of each chiral center.
-
The D-L system refers to a relative configuration of the molecule comparing the molecule to glyceraldehyde enantiomers, used as the standard reference compound.
-
Drugs that have the same relative configuration as (+)-glyceraldehyde use the D prefix; those with the (-)-glyceraldehyde configuration are given the L prefix.
-
-
-
For example, l-propranalol is about two orders of magnitude (100x) then the dextrorotary form.
-
Although timolol is an example of a drug in which only the single L-isomer is used in the formulation, most β-blockers are marketed as the racemate (50:50).
-
However, if another physiological parameter is the endpoint, the D-propranalol inhibits conversion of thyroxine (T4) to triiodothyronine (T3) an effect not observed with the L-form.
-
This example
and others suggest that when dealing with a drug
used as the racemate, each enantiomer exhibits
particular pharmacological profiles that can be
different, similar, opposite or without a
difference between them.
-
-
The calcium channel blockers, which have several cardiovascular indications, are used as racemates.
-
The ACE (angiotensin converting enzyme) inhibitors are chiral compounds with most of them marketed as the single L-isomer.
-
Valsartin is an example of an angiotensin II receptor blocker is administered as a single isomer.11
-
-
-
-
-
|
|
|
|
|
|
|
|