|
|
|
|
|
|
|
-
"P450 Enzyme System (Inducers, Inhibitors & Subtypes")
-
Attribution:
-
Dirty Medicine P450 Enzyme System (Inducers, Inhibitors, & Subtypes) 3/16/2019.
-
Dirty Medicine Youtube Channel https://www.youtube.com/channel/UCZaDAUF7UEcRXIFvGZu3O9Q
-
-
-
Cytochrome P450 Enzyme Induction:
-
Following repeated administration, some drugs increase the amount of P450 enzyme usually by:
-
Increase enzyme synthesis rate (induction)
-
Reduced enzyme degradation rate
-
-
-
Cytochrome P450 enzyme inhibition:
-
Certain drugs, by binding to the cytochrome component, act to competitively inhibit metabolism. Examples:
-
Cimetidine (Tagamet) (anti-ulcer --H2 receptor blocker) and Ketoconazole (Nizoral) (antifungal) bind to the heme iron a cytochrome P450, reducing the metabolism of:
-
Testosterone
-
Other coadministered drugs
-
Mechanism of Action: competitive inhibition
-
-
-
Catalytic inactivation of cytochrome P450.
-
Macrolide antibiotics (troleandomycin, erythromycin estolate (Ilosone)), metabolized by a cytochrome P450:
-
Metabolites complex with cytochrome heme-iron: producing a complex that is catalytically inactive.
-
-
Chloramphenicol (Chloromycetin): metabolized by cytochrome P450 to an alkylating metabolite that inactivates cytochrome P450
-
Other inactivators: Mechanism of Action: -- targeting the heme moiety:
-
Steroids:
-
Ethinyl estradiol (Estinyl)
-
Norethindrone (Aygestin)
-
Spironolactone (Aldactone)
-
-
Others:
-
Propylthiouracil
-
Ethchlorvynol (Placidyl)
-
-
-
-
-
-
Some Phase II Reactions
Type of Conjugation
Endogenous Reactant
Transferase (Location)
Types of Substrates
Examples
Glucuronidation
UDP glucuronic acid
UDP glucuronosyl transferase (microsomal)
phenols, alcohols, carboxylic acids, hydroxylamines, sulfonamides
morphine, acetaminophen, diazepam, digitoxin, digoxin, meprobamate
Acetylation
Acetyl-CoA
N-Acetyl transferase (cytosol)
Amines
Sulfonamides, isoniazid, clonazepam, dapsone, mescaline
Glutathione conjugation
Glutathione
GSH-S-transferase (cytosolic, microsomes)
Epoxides, nitro groups, hydroxylamines
Ethycrinic acid, bromobenzene
Sulfate conjugation
Phosphoadenosyl phosphosulfate
Sulfotransferase (cytosol)
Phenols, alcohols, aromatic amines
Estrone, 3-hydroxy coumarin, acetaminophen, methyldopa
Methylation
S-Adenosyl-methionine
Transmethylases (cytosol)
Catecholamines, phenols, amines, histamine
Dopamine, epinephrine, histamine, thiouracil, pyridine
-
Adapted from Table 4-3, Correia, M.A., Drug Biotransformation. in Basic and Clinical Pharmacology, (Katzung, B. G., ed) Appleton-Lange, 1998, p 57.
-
-
-
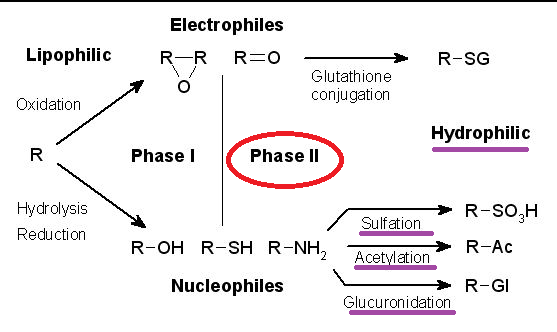
Overview: Phase II reactions involve non-microsomal enzymes
-
Xenobiotic Metabolism
-
Attribution:
-
TimVickers, Public domain via Wikimedia Commons
-
https://commons.wikimedia.org/wiki/File:Xenobiotic_metabolism.png
-
https://upload.wikimedia.org/wikipedia/commons/9/97/Xenobiotic_metabolism.png
-
-
-
 Reaction types:
Reaction types:-
Conjugation
-
Hydrolysis
-
Oxidation
-
Reduction
-
Location (non-microsomal enzymes): primarily hepatic (liver); also plasma and gastrointestinal tract
-
Non-microsomal enzymes catalyze all conjugation reactions except glucuronidation
-
-
-
Nonspecific esterases in liver, plasma, gastrointestinal tract hydrolyzed drugs containing ester linkages, including
-
Succinylcholine (Anectine),
-
Atricurium (Tracrium),
-
Mivacurium (Mivacron),
-
Esmolol (Brevibloc) as well as ester-type local anesthetics.
-
-
Conjugation reactions are usually "detoxification reaction".
-
Conjugates tend to be more polar compared to the parent compound, more easily excreted, and usually pharmacologically inactive.
-
-
Conjugation reactions require "high-energy" intermediates in specific transfer enzymes which include both microsomal and cytosolic transferases.
-
Conjugation with glucuronic acid:
-
Glucuronic acid is available from glucose and its conjugation with lipid-soluble drugs results in a lipophilic glucuronic acid derivative which is typically pharmacologically inactive and more water-soluble compared to the parent compound.
-
Therefore, the glucuronic acid derivative molecule is more readily excreted in both urine or bile.
-
-
-
Transferases are enzymes which catalyzes the coupling of an endogenous substance with the drug.
-
For example, transferase which catalyzes the "transfer" of uridine-5'-diphosphate (UDP) derivative of glucuronic acid and a drug.
-
A transferase may catalyze an inactivated drug with an endogenous substrate. For example a S-CoA derivative of benzoic acid with an endogenous substrate.
-
-
-
-
-
-
Certain conjugation reactions form toxic reactive species (hepatotoxicity). For example, acyl glucuronidation of nonsteroidal anti-inflammatory drugs may result in toxicity. Another example would be N-acetylation of isoniazid.
-
Drugs metabolized to toxic products:
-
Acetaminophen hepatotoxicity (normally safe in therapeutic doses)
-
Therapeutic doses:
-
Glucuronidation + sulfation to conjugates (95% of excreted metabolites); 5% due to alternative cytochrome P450 depending glutathione (GSH) conjugation pathway
-
-
 At high doses:
At high doses:-
Glucuronidation and sulfation pathways become saturated
-
Cytochrome P450 dependent pathway becomes now more important.With depletion of hepatic glutathione, hepatotoxic, reactive, electrophilic metabolites are formed.
-
In this circumstance antidotes would include N-acetylcysteine and cysteamine. N-acetylcysteine protects patients from fulminant hepatotoxicity and death following acetaminophen overdose.
-
-
-
-
Stoelting, R.K., "Pharmacokinetics and Pharmacodynamics of Injected and Inhaled Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, 1-17.
Benet, Leslie Z, Kroetz, Deanna L. and Sheiner, Lewis B The Dynamics of Drug Absorption, Distribution and Elimination. In, Goodman and Gillman's The Pharmacologial Basis of Therapeutics,(Hardman, J.G, Limbird, L.E, Molinoff, P.B., Ruddon, R.W, and Gilman, A.G.,eds) TheMcGraw-Hill Companies, Inc.,1996, pp. 3-27
Correia, M.A., Drug Biotransformation. in Basic and Clinical Pharmacology, (Katzung, B. G., ed) Appleton-Lange, 1998, pp 50-61
-
Basis for individual to individual variation in drug responses
-
Response Variation Secondary to Pharmacokinetic Differences
-
Bioavailability
-
Renal function
-
Liver function
-
Cardiac function
-
Patient Age
-
-
Response Variation Secondary to Pharmacodynamic Differences
-
Enzyme activity
-
Genetic differences
-
-
Response Variation Secondary to Drug Interactions
-
Stoelting, R.K., "Pharmacokinetics and Pharmacodynamics of Injected and Inhaled Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, 1-17.
-
Genetic Factors: in Biotransformation of Drugs
-
Genetic influences: Variation in drug metabolism rates or in receptor sensitivity:
-
Metabolism:
-
Patients can be categorized as either rapid or slow acetylators; a classification which refers to the patients ability to relatively rapidly or slowly catalyze acetylation reactions.
-
Biotransformation of some drugs are affected by acetylation rates, examples include hydralazine (Apresoline) and isoniazid (INH).:
-
-
-
Pharmacogenetics: One major concern is that on underlying disease state may not be appreciated until an unexpected reaction to an anesthetic agent in fact occurs. The anesthetic agent essentially exposes on underlying disease state and then appropriate inner operative responses required.
-
Examples:
-
Atypical cholinesterase enzyme suggested by prolonged succinylcholine (Anectine) or mivacurium (Mivacron)-induced neuromuscular blockade
-
Succinylcholine (Anectine) or volatile anesthetic induced malignant hyperthermia:
-
Malignant hyperthermia is a very serious reaction requiring a definitive treatment approach including dantrolene (Dantrium).
-
-
If the patient exhibits glucose-6-phosphate dehydrogenase deficiency certain drugs may induce hemolysis
-
Barbiturates may induce intermittent porphyria attacks. It is extremely important to determine therefore preoperatively if the patient has history of intermittent porphyria.
-
-
-
|
-
Influence of Age on
Drug Responses
-
Variation in drug responses may be due to several factors such as:
-
Diminished cardiac output:
-
A reduction in cardiac output reduces hepatic perfusion which may decrease delivery of drug to the liver for metabolism. This type of an effect would prolonged duration of action of, for example, lidocaine (Xylocaine) or fentanyl (Sublimaze).
-
-
Increased body fat:
-
An increase in body fat tends to increase Vd .
-
An increased Vd would tend to prolong clearance time.
-
-
Increased body fat also promotes accumulation of highly lipid-soluble agents such as diazepam (Valium) and thiopental (Pentothal).
-
-
Altered protein binding can affect drug responses because only the "free", unbound drug is active and for a highly protein-bound drug small changes in the extent of protein binding can substantially influence the free drug concentration [free drug].
-
Decreased or compromised renal function can prolong drug action if renal excretion is the primary mechanism for clearance.
-
-
Stoelting, R.K., "Pharmacokinetics and Pharmacodynamics of Injected and Inhaled Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, 1-17.
-
-
Definition: Drug interaction -- when one drug affects the pharmacological response of a second drug given at the same time.
-
Drug interactions may be due to:
-
Pharmacodynamic effects
-
Pharmacokinetic effects
-
-
Consequences of drug interactions:
-
Increased drug effects; decreased drug effects
-
Desired consequences; adverse or undesired effects
-
-
Examples -- positive, beneficial drug interaction effects:
-
Propranolol + hydralazine (reflex tachycardia (undesirable) caused by hypotensive hydralazine-mediated response is prevented by propranolol-mediated β-adrenergic receptor blockade
-
Opioid-induced respiratory depression may be counteracted by administration of the opioid receptor antagonist naloxone
-
-
Adverse effects -- toxic reactions
-
One drug may interact with another to impede absorption
-
One drug may compete with another for the same plasma protein-binding sites
-
One drug may affect metabolism of another by either enzyme induction or enzyme inhibition
-
One drug may change the renal excretion rate of the other.
-
-
Stoelting, R.K., "Pharmacokinetics and Pharmacodynamics of Injected and Inhaled Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, 1-17.
Dolin, S. J. "Drugs and pharmacology" in Total Intravenous Anesthesia, pp. 13-35 (Nicholas L. Padfield, ed), Butterworth Heinemann, Oxford, 2000
|
|
|
|
|
|
|