|
|
|
|
|
|
|
Medical Pharmacology Chapter 33-34: Anticancer Drugs
|
|
|
|
|
|
|
Antimetabolites
Purine Analogues:
Nelarabine (Arranon)
|
|
|
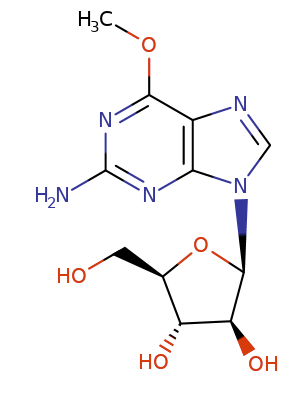
Nelarabine is an FDA approved purine analog (2005).1,8
![]() Its
clinical indication is for treatment of T-cell acute leukemia (T-ALL) and
T-cell lymphoblastic lymphoma (T-LBL) not responding (or relapsed) after a
minimum of two previous chemotherapy protocols.
Its
clinical indication is for treatment of T-cell acute leukemia (T-ALL) and
T-cell lymphoblastic lymphoma (T-LBL) not responding (or relapsed) after a
minimum of two previous chemotherapy protocols.
|
|
Nelarabine (6-methoxy-arabinosyl-guanine) has the distinction of being the singular guanine nucleotide use clinically.1
When used in the treatment of acute T-cell leukemia about 1/5 (20%) of patients exhibits complete responses.8
The underlying mechanism of action is similar to previous purine analogs that require activation by sequential phosphorylation steps.
![]() Ultimately, ara-G
upon phosphorylation to ara-GTP is incorporated into DNA, thereby
terminating DNA elongation resulting in DNA synthesis inhibition and cell
death.8
Ultimately, ara-G
upon phosphorylation to ara-GTP is incorporated into DNA, thereby
terminating DNA elongation resulting in DNA synthesis inhibition and cell
death.8
DNA synthesis is inhibited with increasing concentrations of intracellular deoxyguanosine.
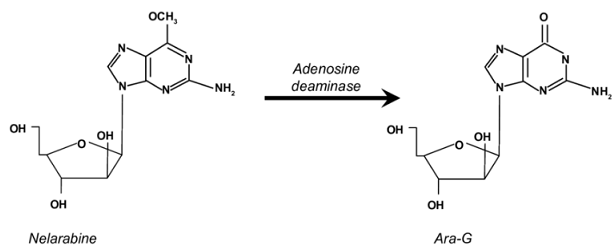
However, the short half-life (t1/2) of plasma deoxyguanosine, due to rapid deamination catalyzed by purine nucleoside phosphorylase (PNP) highly concentrated in erythrocytes, make this agent unsuitable as a drug.
Ara-G (9-β-D arabinofuranosyl guanine), an analog of deoxyguanosine resists PNP catalyzed deamination but exhibits limited solubility.
![]() Nelarabine is
related to ara-G given that it is a soluble prodrug, due to its
6-O-methylated moiety.
Nelarabine is
related to ara-G given that it is a soluble prodrug, due to its
6-O-methylated moiety.
Therefore, nelarabine is a prodrug of ara-G and is converted to ara-G by the action of the enzyme adenosine deaminase (ADA).
|
|
Ara-G is phosphorylated to ara-GTP using to enzymes, deoxycytidine kinase and deoxyguanosine kinase.
![]() Ara-GTP upon
incorporation into DNA results in termination of DNA elongation and
subsequently to DNA synthesis inhibition and cell death.
Ara-GTP upon
incorporation into DNA results in termination of DNA elongation and
subsequently to DNA synthesis inhibition and cell death.
Depletion of deoxynucleotides needed for DNA synthesis is also mediated by ara-GTP through inhibition of ribonucleotide reductase (RNR).
 |
|
The likelihood of a clinical response to nelarabine treatment is significantly dependent on adequate nelarabine uptake and significant accumulation of ara-GTP by the target leukemic cells.
Absorption, Distribution, Biotransformation, Excretion:1
Nelarabine is administered by the intravenous route of administration.1
Nelarabine, a prodrug, must be activated for realization of his neoplastic activity.
The initial step in activation is an enzyme catalyzed removal of a methyl group resulting in ara-G which is phosphorylase resistant.
The half-life (plasma t1/2) of ara-G is about 3 hours.
Ara-G is transported into tumor cells and then activated by deoxycytidine kinase and deoxyguanosine kinase to a triphosphate form, ara-GTP.
This metabolite is incorporated into DNA, terminating its synthesis.
Nelarabine as well as ara-G are mainly eliminated by metabolism to guanine.
Renal excretion is responsible for a small elimination fraction; however, in cases of significant renal dysfunction, nelarabine administration should be accompanied by careful clinical assessments.1
The major nelarabine toxicities are associated with central nervous system (CNS) effects.
These effects include:
Seizures
Encepalopathy
Obtundation (reduced alertness) and
Peripheral neurotoxicity.
Uncommonly, ascending myelopathy may occur exhibiting sensory and motor loss along with posterior column changes noted on MRI imaging.
With respect to most common adverse effects, malaise, headache, somnolence, nausea, fever, peripheralneuropathy and myelosuppression have been reported.
Notable neurologic events (grade 3 or 4) appear to occur with a frequency of about 10% to 15%.8
The primary clinical use for nelarabine is in the treatment of individuals with acute lymphoblastic leukemia and lymphoblastic lymphoma (LBL).
In the case of patients exhibiting relapsed or refractory T-cell malignant disease, response rates are relatively low, ranging from about 20% in pediatric patients to about 30% in adults.
Some patients may be candidates for stem cell transplantation treatment.8
|
|
|
|
|
|
|