![]()
![]()
|
|
|
Anesthesia Pharmacology Chapter 28: Pain Management
![]()
![]()
|
|
|
|
|
Clinical Use: Opioid Analgesics
|
Opioids
"Narcotic" -- imprecise term suggesting "narcosis": indicated of a somnolent state
"Opioid analgesic" -- more appropriate: suggesting analgesia (pain absence)
without resulting in loss of consciousness/sleep
all natural/semisynthetic opium alkaloid derivatives
synthetic agents
other drugs whose opioid-like effects are blocked by naloxone (Narcan) -nonselective opioid receptor antagonist
Opium -- from the opium poppy
Constituents:
morphine
codeine
thebaine-- nonanalgesic
papaverine-- nonanalgesic, vasodilator
full agonists (high efficacy, strong agonists)
partial agonists
may cause agonist effects; may displace full agonists thereby reducing their effects.
a partial agonist may act either as an agonist or antagonist
antagonists: multiple opioid receptor subtypes allow agonists/antagonist combination effects -- e.g.:
opioid -- agonist at one opioid receptor subtype; partial agonist or antagonist at another subtype -- "agonist-antagonists" or "mixed agonist-antagonists"
Examples:
naloxone (Narcan): pure antagonist: no effects normally associated with agonist binding
morphine: full agonist at mu receptor
codeine: partial or "weak" agonist -- < maximal effect with complete receptor saturation
nalbuphine (Nubain) : agonist that one opioid receptor; antagonist at another
Partial agonist/antagonist characteristics: replacement of methyl moiety on the nitrogen atom with larger substituents:
Allyl substitution-- nalorphine and naloxone
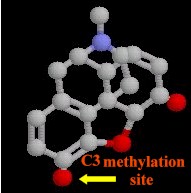
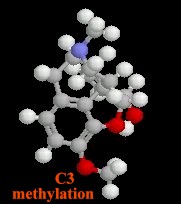
Substitutions at the C3 and C6 morphine hydroxyl groups (see below)
Pharmacokinetic properties altered
Methyl substitution at C3 reduces first-pass hepatic metabolism by glucuronide conjugation: -- as a consequence codeine and oxycodone have a higher oral: parenteral potency

|
|
|
|
|
Acetylation of both morphine hydroxyls = heroin {more rapid access across the blood-brain barrier compared morphine}; in the brain heroin:
rapidly hydrolyzed to monoacetylmorphine and morphine
Morphine (and related agents) cause analgesia by acting the brain regions containing peptides which have opioid-like properties
Endogenous substances = endogenous opioid peptides
Previous used term "endorphin" now refers to ß-endorphins and related peptides derived from the precursor: prepro-opiomelanocortin
Most widely distributed opioid analgesic peptides:
pentapeptides
methionine-enkephalin (met-enkephalin)
leucine-enkephalin (leu-enkephalin)
Three major precursor proteins:
prepro-opiomelanocortin (POMC) {contains}:
met-enkephalin sequence
ß-endorphin sequence
some nonopioid peptides:
ACTH
ß-lipotropin
melanocyte-stimulating hormone
preproenkephalin (proenkephalin A ) {contains}:
six copies of met-enkephalin
one copy of leu-enkephalin
preprodynorphin (proenkephalin B) {contains-- active peptides containing the leu-enkephalin sequence}:
dynorphin A
dynorphin B
a and ß neoendorphin
Endogenous opioid precursors: localized at pain modulation brain regions
May be released during: stress (pain; pain anticipation)
Precursor molecules also found:
adrenal medulla
neural plexuses of the gut
opioid analgesics: generally well absorbed destined cutaneous/intramuscular/mucosal surfaces
fentanyl transdermal: important Route of Administration
Gastrointestinal absorption:
some opioids-- subject to first-pass effects:
codeine; oxycodone -- high oral: parenteral potency (protected from conjugation by substitution on C3 aromatic hydroxyl)
various extent of plasma protein binding
highest concentrations in tissues: function of perfusion
skeletal muscle: largest reservoir
for highly lipophilic opioids(e.g. fentanyl): concentration in adipose tissue
Blood Brain Barrier:
amphoteric agents (possessing both an acidic and basic group, e.g. morphine {phenolic hydroxyl at C3}: greatest difficulty for brain entry)
other substitutions that C3 improve blood-brain barrier penetration: e.g., heroin, codeine
neonatal considerations: neonates lack the blood-brain barrier:
placental opioid transfer (uses in obstetric analgesia) can result in depressed respiration in the newborn.
Conversion to polar metabolites; renal excretion
Opioids with hydroxyl groups: likely conjugated with glucuronic acid
Examples: morphine , levorphanol (Levo-dromoran)
morphine-6-glucuronide: analgesic potency (perhaps > parent compound morphine)
in patients with compromised renal function:accumulation metabolites -- prolonged analgesia
Esters: hydrolyzed by tissue esterases:
Examples: heroin, remifentanil (short duration of action)
N-demethylation: minor pathway
accumulation of demethylated meperidine (Demerol) metabolite, normeperidine:
patients with decreased renal function or on high dosages: CNS excitatory effects:
seizures (more likely in children)
Oxidative metabolism (hepatic) primary route of phenylpiperidine opioid metabolism:
fentanyl (Sublimaze)
alfentanil (Alfenta)
sufentanil (Sufenta)
polar metabolites -- renal; small amounts excreted unchanged
glucuronide conjugates -- bile (enterohepatic circulation minor)
|
Way, W.L., Fields, H.L. and Way, E. L. Opioid Analgesics and Antagonists, in Basic and Clinical Pharmacology, (Katzung, B. G., ed) Appleton-Lange, 1998, pp 496-515. |
|
Coda, B.A. Opioids, In Clinical Anesthesia, 3rd Edition (Barash, P.G., Cullen, B.F. and Stoelting, R.K.,eds) Lippincott-Ravin Publishers, Philadelphia, New York, 1997, pp 329-358. |
|
Schuckit, M.A. and Segal D.S., Opioid Drug Abuse and Dependence, In Harrison's Principles of Internal Medicine 14th edition, (Isselbacher, K.J., Braunwald, E., Wilson, J.D., Martin, J.B., Fauci, A.S. and Kasper, D.L., eds) McGraw-Hill, Inc (Health Professions Division), 1998, pp 2508-2512. |
analgesia: specific receptor binding -- localization:
spinal cord
brain
Mu (μ)
Delta (δ)
Kappa (κ)
General Opioid Receptor Characteristics:
G protein coupled receptor family
Significant amino acid sequence homology
Each-receptor: subtypes
μ1, μ2
δ1, δ2
κ1, κ2
Receptor types and physiological effects:
Mu (m) :Analgesia, euphoria, respiratory depression, physiological dependence
Most opioid analgesics: act at the mu receptor
Delta (d) and Kappa (k): Spinal analgesia
Drugs/endogenous opioids: Receptor- type affinity
morphine -- (μ)
pentazocine -- (κ) some (μ)
endogenous opioid peptides:
leu-enkephalin --(δ)
dynorphin --(κ)
|
Drug |
Mu (m) |
Delta (d) |
Kappa (k) |
|
Opioid Peptides |
|||
|
Enkephalins |
Antagonist |
Agonist |
|
|
beta-endorphin |
Agonist |
Agonist |
|
|
Dynorphin |
Weak Agonist |
Agonist |
|
|
Agonists |
|||
|
Codeine |
Weak Agonist |
Weak Agonist |
|
|
etorphine |
Agonist |
Agonist |
Agonist |
|
fentanyl (Sublimaze) |
Agonist |
||
|
meperidine (Demerol) |
Agonist |
||
|
methadone (Dolophine) |
Agonist |
||
|
Morphine |
Agonist |
Weak Agonist |
|
|
Agonist-antagonists |
|||
|
Buprenorphine |
Partial Agonist |
||
|
dezocine (Dalgan) |
Partial Agonist |
Agonist |
|
|
nalbuphine (Nubain) |
Antagonist |
Agonist |
|
|
pentazocine (Talwain) |
Antagonist or Partial Agonist |
Agonist |
|
|
Antagonist: naloxone (Narcan) |
Antagonist |
Antagonist |
Antagonist |
Opioids:G protein linked-- affecting
ion channel state
intracellular Ca2+ levels
protein phosphorylations states
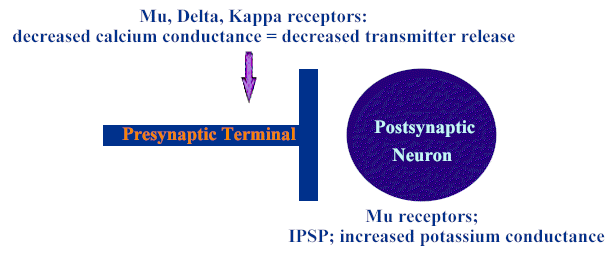
Two well-defined opioid actions:
reduce neurotransmitter release; by closing a voltage-gated Ca2+ channel on presynaptic neuronal terminals Or
inhibit postsynaptic neurons (hyperpolarization) by increasing and K+ channel conductance
Spinal cord presynaptic sites:
reduced transmitter released-- affects acetylcholine, norepinephrine, glutamate, serotonin, substance P
|
|
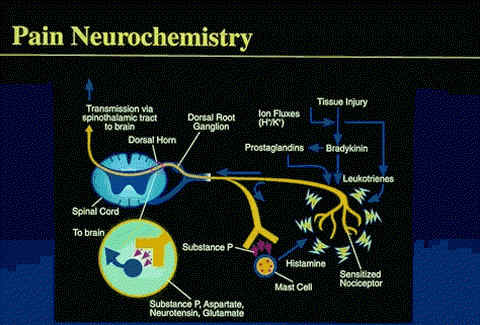
Serotonin, bradykinin, histamine, prostaglandins, substance P (sP) , and various ions (ie, H+ or K+)--the biochemical mediators released as a result of tissue injury--have been implicated in nociceptive activation and sensitization (hyperalgesia).
Hyperalgesia results in enhancement of spontaneous pain via a reduction in pain threshold and a lengthening in duration of nociceptor response to stimuli.
PGE1, PGE2, and PGF2a, are the most potent prostaglandins to produce these sensitization effects.
Substance P, synthesized by cells of the spinal ganglia, has been identified at the peripheral terminal of unmyelinated primary afferent fibers.
This putative neurotransmitter may play a role in the propagation of visceral nociceptive pain from the gastrointestinal (GI) tract, ureters, and urinary bladder.
In addition, to sP, other potential nociceptive transmitters include glutamate, aspartate, somatostatin, cholecystokinin, and vasoactive intestinal polypeptide.
courtesy of Roxane Pain Institute used with permission
|
Way, W.L., Fields, H.L. and Way, E. L. Opioid Analgesics and Antagonists, in Basic and Clinical Pharmacology, (Katzung, B. G., ed) Appleton-Lange, 1998, pp 496-515. |
|
Schuckit, M.A. and Segal D.S., Opioid Drug Abuse and Dependence, In Harrison's Principles of Internal Medicine 14th edition, (Isselbacher, K.J., Braunwald, E., Wilson, J.D., Martin, J.B., Fauci, A.S. and Kasper, D.L., eds) McGraw-Hill, Inc (Health Professions Division), 1998, pp 2508-2512. |
|
Coda, B.A. Opioids, In Clinical Anesthesia, 3rd Edition (Barash, P.G., Cullen, B.F. and Stoelting, R.K.,eds) Lippincott-Ravin Publishers, Philadelphia, New York, 1997, pp 329-358. |
primary afferents to pain transmission neurons
Opioid agonists:
inhibit excitatory transmitters release from these primary afferents
inhibit dorsal horn pain transmission neurons
Clinical application: directed demonstration of opioid agonists allow regional analgesia which minimizes CNS side effects
Systemic Opioid Administration:
Important opioid binding sites in descending pathways
rostral ventral medulla
locus ceruleus
midbrain periaqueductal gray
Administration of exogenous opioids promotes release of endogenous opioids
Repeated opioid administration:
gradual loss of effect, e.g. tolerance
Physical Dependence = physiological withdrawal symptoms (abstinence syndrome) if an antagonist is administered or the agonist is stopped.
Tolerance is not developed equally to all opioid effects.
|
|
|
|
|
|
|
|
|
![]()
![]()
|
|
|