|
|
|
Autonomic Pharmacology--Adrenergic Drugs
|
|
|
|
|
|
|
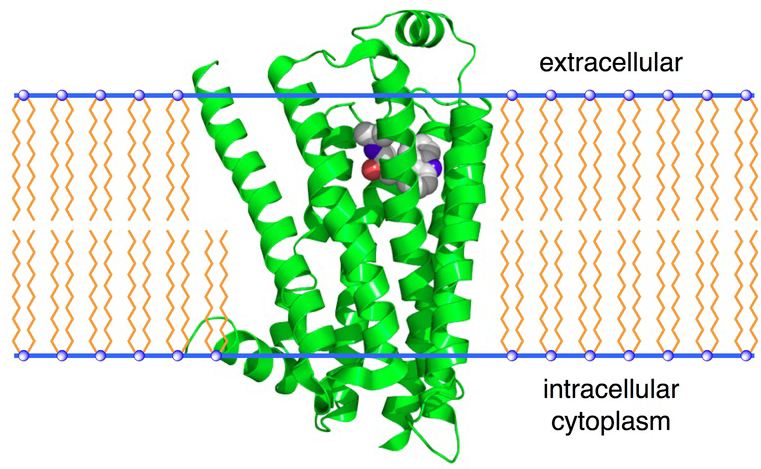
→β2-Adrenergic Receptors←
Adrenergic Receptor Subtypes continued:
β2-adrenergic receptors have been identified in liver, skeletal muscle, and in smooth muscle.9
In smooth muscle, activation of this β-adrenergic receptor subtype results in stimulation (or activation) of Gs, increased adenylyl cyclase activity, increased [cAMP], and increased protein kinase A activity.
Recall that kinases phosphorylate target sites and in this case, increasing protein kinase A catalytic activity promotes phosphorylation of a protein important in smooth muscle contraction and relaxation, myosin light chain kinase.
Increased phosphorylation of myosin light chain kinase reduces its tendency to bind calcium-calmodulin and this favors smooth muscle relaxation. Dephosphorylation reactions are catalyzed by phosphatases and balance the actions of kinases in regulation of target site phosphorylation state.
Sometimes one associates β2-adrenergic agonists with treatment for asthma. Relaxation of bronchial smooth muscle, mediated by β2-adrenergic receptors, may proceed by a Gs-independent increase in K+ channel conductance. In this model, increasing K+ conductance enhances movement of potassium ions out of the cell. Having fewer positive charged ions inside the cell results in membrane hyperpolarization, i.e. movement of the membrane potential to more negative potentials, thus opposing membrane depolarization. Depolarization itself is associated with contraction; therefore, membrane hyperpolarization b tends to reduce bronchiolar smooth muscle tone.
Activation of β2-adrenergic receptors in the liver utilizes the Gs signaling system which, through subsequent phosphorylation steps, increases catalytic activity of glycogen phosphorylase and glycogen catabolism. One result of this activity is increased plasma glucose. For skeletal muscle, the same process stimulates both glycogenolysis and K+ uptake.9
β2-adrenergic receptor activation sometimes is associated with the Gq G protein subunit. This association results multiple kinase activations which influence many intracellular signaling pathways.35
Subdivision of β-adrenergic receptors reflects potency differences of various adrenergic agonists.46
For example, the subdivision of β receptors into β1 and β2-adrenergic receptor subtypes with β1 (myocardial) and β2 (smooth muscle and most other locations) differing with respect to epinephrine and norepinephrine potencies.46
That is, epinephrine and norepinephrine exhibit similar potencies at β1; however, epinephrine is about 10-50 times more potent at β2 receptors.46
Differences between adrenergic receptors are also reflected in antagonist affinities. 46
 |
|
The existence of a third β-adrenergic receptor subtype, β3, was suggested by "atypical" responses to adrenergic receptor antagonists, i.e. the β3-receptor subtype exhibited low affinity.47
Prior to definitive identification of this receptor subtype, the idea of other adrenergic receptor types beyond β1 and β2 had been proposed to account for sympathetic regulation of metabolic processes in the digestive tract, adipose tissue and skeletal muscle.47
Also, in the 1980s adrenergic receptor agonists were developed that caused metabolic rate stimulation, thermogenesis in adipose tissue, ileus relaxation, and soleus muscle glycogen synthesis. These agonists had limited action at β1 and β2 adrenergic receptors, however. In 1989, a human gene was isolated that encoded the third β-adrenergic receptor, β3.47
Pharmacological properties were investigated in addition to tissue distribution for the β3 adrenergic receptor. 47
In cultured cells expressing the β3 subtype (no expression of β1 or β2), most classical β-adrenergic antagonists were unable to inhibit isoproterenol-induced cAMP accumulation. The β3 adrenergic receptor is about 10 times more sensitive to norepinephrine and epinephrine.
Adipocytes represent a primary location for β3 receptor localization, although all of the β receptor subtypes are present in white and brown adipose tissue.46
Following administration of β3-receptor agonists, both lipolysis and thermogenic responses are observed in animals.46
Small variations in the β3 receptors gene (genetic polymorphism) might be a factor in susceptibility to type 2 diabetes and obesity risk.46
One study considered the time of onset of non-insulin-dependent diabetes mellitus (NIDDM) and genetic variation in the β3 receptor gene. 47
The gene was analyzed in 10 Pima Indians with assessment of single-stranded conformational polymorphism and dideoxy sequence analysis. Also, association studies in 642 Pima individuals (390 with NIDDM and 252 without NIDDM) were undertaken. 47
A "missense" mutation in the β3-adrenergic receptor gene resulted in the substitution of a tryptophan by arginine in the first intracellular loop of the receptor protein. 47
β3-Adrenergic Receptor Protein Diagram47 Each amino acid is shown as a circle. The substitution of a tryptophan by arginine (due to a missense mutation) is illustrated at the beginning of the first intracellular loop.47 In those subjects homogeneous for this mutation the onset of NIDDM was occurred at an earlier age.47
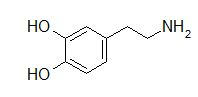
Dopamine is an important endogenous agent that acts by interacting not only with dopamine receptors (D1) but also with multiple subtypes of catecholamine receptors in the periphery.9 (Other dopamine receptor types (D2-like) will be discussed later).
 |
 |
The predominant receptor-mediated effect will depend on the concentration of dopamine infused. This consequence follows in part from differences in affinities between dopamine and various receptors.
Accordingly, at low dopamine concentrations the first receptors to be increasingly saturated and activated will be those that exhibit the highest dopamine-receptor affinity. As the dopamine concentration increases other receptor types will be increasingly occupied and effects mediated by activation of those receptors become prominent.
For example, at "low dose" dopamine (<2 μg/kg/min infusion rate) the predominant dopamine effect is mediated by D1 dopaminergic receptors in the following vascular beds:
mesentery
renal
coronary. 9
A principal physiological consequence of D1 receptor activation is vasodilatation.
Vasodilatation occurs subsequent to D1 receptor activation which increases adenylyl cyclase activity in vascular smooth muscle that in turn increases [cAMP].9
However, at higher infusion rates (2-10 μg/kg/min), dopamine exhibits positive inotropism (increased myocardial contractility) by activation of β1 adrenergic receptors.
Even higher dopamine concentrations, seen at increased rates of infusion (>10 μg/kg/min), vasoconstriction is observed due to dopamine-activation of α1 receptors.9
The sequence of D1, β1, α1 receptor activation illustrates that one aspect of drug selectivity is concentration dependent.
In this example increasing likelihood of first D1, then β1 and finally α1 receptor activation is dopamine concentration dependent; however, another type of progression, from desired therapeutic pharmacological actions of a drug to adverse effects of a drug may also progress in accordance with drug concentrations. Firstly, the therapeutically-targeted receptor is activated and then, at higher drug concentrations, the drug activates other sites associated with undesirable effects.
![]()
|
|
|
|
|
|
|
|
| This Web-based pharmacology and disease-based integrated teaching site is based on reference materials, that are believed reliable and consistent with standards accepted at the time of development. Possibility of human error and on-going research and development in medical sciences do not allow assurance that the information contained herein is in every respect accurate or complete. Users should confirm the information contained herein with other sources. This site should only be considered as a teaching aid for undergraduate and graduate biomedical education and is intended only as a teaching site. Information contained here should not be used for patient management and should not be used as a substitute for consultation with practicing medical professionals. Users of this website should check the product information sheet included in the package of any drug they plan to administer to be certain that the information contained in this site is accurate and that changes have not been made in the recommended dose or in the contraindications for administration. Advertisements that appear on this site are not reviewed for content accuracy and it is the responsibility of users of this website to make individual assessments concerning this information. Medical or other information thus obtained should not be used as a substitute for consultation with practicing medical or scientific or other professionals. |
