|
|
|
|
|
|
|
Nursing Pharmacology Chapter 33-34: Anticancer Drugs
|
|
|
|
|
|
|
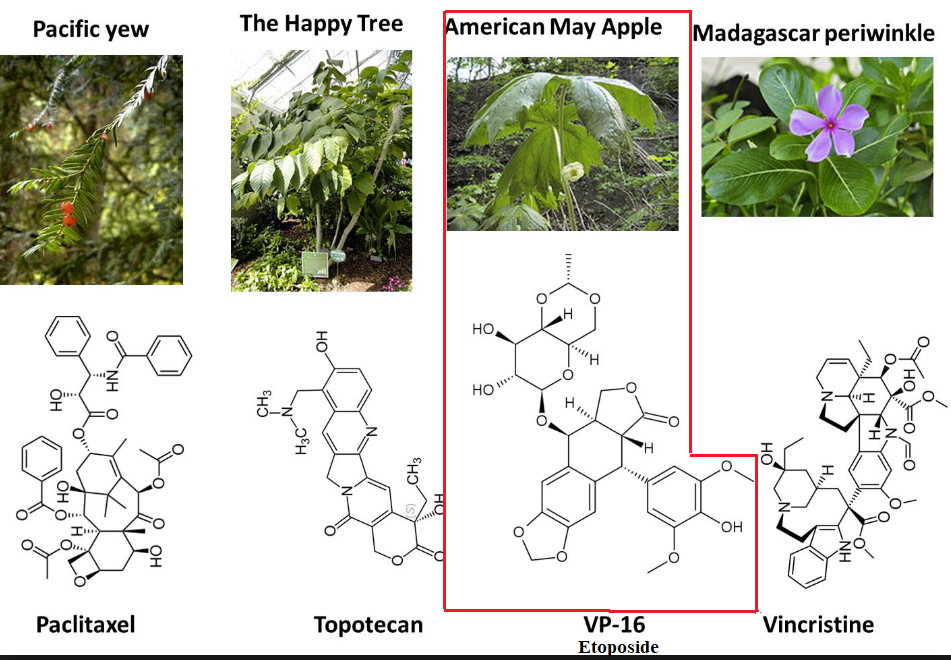
Natural Products:
Topoisomerase Inhibitors Type II: Epipodophyllotoxins: Etoposide (VP-16, Etopophos, Toposar)
|
|
 |
|
The mayapple or mandrake plant root extracts have been a source of medicinals.
|
|
|
 |
|
The active agent, podophyllotoxin, exerts its antineoplastic action as an anti-mitotic agent binding to tubulin.
Podophyllotoxin binds at a location that differs from that associated with tubulin.
Several semisynthetic podophyllotoxin derivatives have been synthesized, two of which are neoplastic agents which target topoisomerase II.
![]() The two
demethylepipodophyllotoxin drugs used in cancer treatment are etoposide
(VP-16) and teniposide.
The two
demethylepipodophyllotoxin drugs used in cancer treatment are etoposide
(VP-16) and teniposide.
Absorption, Distribution, Biotransformation, Excretion:8,9
Etoposide is administered by the IV or oral route of administration.8,9
Following an IV dose, etoposide elimination is best modeled by a two-compartment pharmacokinetic approach.
The terminal half-life is about 7 hours (range = 4-11 hours) for adult patients with normal kidney function.
However, interpatient differences with respect to pharmacokinetic factors exhibit a range of about 35%.
For oral administration, notable inter-and intrapatient variation have been described.
Half-life of elimination in children is also about 7 hours with the range of 6 to 8 hours.
Bioavailability following oral administration approximates 50% with the range described as 25% to 75%.
Oral administration may be particularly beneficial in order to obtain clinically relevant drug concentrations over extended periods of time.
The effectiveness of oral etoposide treatment depends on managing both pharmacokinetic issues such as variable and low bioavailability.
Oral bioavailability at doses greater than about 200 mg is subject to saturation, likely is saturable absorption process in the G.I. tract.
Etoposide is highly bound to plasma proteins, about 96%.8,9
The extent of protein binding which is nonlinear is affected both by individual drug concentration and albumin.
For example, increased free drug concentration is noted in patients with low serum albumin.
![]() Furthermore,
with elevated serum bilirubin, a competitor for albumin binding, free drug
levels also increase.
Furthermore,
with elevated serum bilirubin, a competitor for albumin binding, free drug
levels also increase.
Higher drug levels under these circumstances appear associated with a more pronounced drug toxicity.
High protein binding itself underlies both the relationship between the unbound etoposide fraction and associated, acute toxicity and limited CSF diffusion.
Limited diffusion also applies to plural and ascitic fluids.
The average volume of distribution (Vd) in children is about 10 L/m2.8,9
In adults, Vd ranges from about 7 to 17 L/m2.
Etoposide crosses the blood-brain barrier to a limited degree, with CSF concentrations found to be <5% of drug concentrations found in plasma.
Etoposide metabolism proceeds by means of:
Phase I (hepatic: cytochrome P450 microsomal drug metabolizing system) and
Phase II (conjugation reactions catalyzed by glutathione-S-transferase; glutathione-S tranferase P; UDP-glucuronosyltransferase 1).8,9
Liver cytochrome P450 metabolism is associated mainly with CYP3A4 and CYP3A5 cytochrome P450 isoforms.
Other metabolic pathways depend on catalysis by prostaglandin synthases or myeloperoxidase catalyzed etoposide to O-demethylated products such as catechol, further oxidized to a quinone and quinine.
The O-demethylated product is biologically active against topoisomerase II.
Orthoquinone and semiquinone, etoposide free radicals, may bind covalently to DNA resulting in DNA strand breakage by topoisomerase II-independent processes.
Etoposide excretion depends on both renal and nonrenal mechanisms.8,9
About 40% of administered etoposide is eliminated by the kidney unchanged.
Reduced creatinine clearance may require reduction in etoposide doses.
Biliary excretion represents a route of limited elimination and small changes in liver function may not require dose reduction, assuming kidney function is normal.
However, elevated bilirubin reduces unbound etoposide clearance resulting in elevated hematologic toxicity.
A reduction in etoposide dose (about 50%) may be appropriate should patients exhibit total bilirubin levels of 1.5 to 3.0 mg/dL.
![]() Increased
etoposide clearance may be associated with coadministration of
anticonvulsants, the administration of which induce hepatic enzymes.8,9
Increased
etoposide clearance may be associated with coadministration of
anticonvulsants, the administration of which induce hepatic enzymes.8,9
The major acute toxicity associated with etoposide is myelosuppression, which is the dose-limiting toxicity.8
Other toxicities include nausea and vomiting and alopecia.
With respect to delayed toxicity, etoposide administration is associated with secondary leukemia.
The most frequent secondary malignancy is acute myelogenous leukemia (AML).
This malignancy appears to usually involve the long arm of chromosome on 11 which exhibits a translocation of the MLL (Myeloid-Lymphoid Leukemia) gene.8
The primary FDA approved uses of etoposide are in small cell lung cancer and testicular cancer.9
|
Testicular cancer: etoposide in combination with bleomycin and cisplatin is administered by the IV route of administration for treatment of testicular cancer.1
Up to half of patients with high-risk clinical stage I non-seminomatous germ cell tumors (NSGCT) may have pathological lymph node involvement.
One clinical option in this setting is retroperitoneal lymph node dissection (RPLND), followed by chemotherapy.
As an example, etoposide has been used in this context with bleomycin and cisplatin with a clinical trial suggesting fewer recurrences when comparing RPLND + chemotherapy with RPLND alone.13
Lung Cancer: Lung cancer: in treating small cell lung cancer (SCLC) etoposide is administered in combination with cisplatin.1
Other cancers: etoposide shows anticancer activity in:
Non-Hodgkin's lymphoma
Acute nonlymphocytic leukemia
Kaposi's
sarcoma associated with AIDS.1
|
|
|
|
|
|
|

