|
|
|
Anesthesia Pharmacology Chapter 5: Autonomic Pharmacology: Adrenergic Drugs
|
|
|
|
|
|
|
In considering the cellular or organ-level responses to endogenous or exogenous adrenergic agents, both the number of adrenergic receptors present and the particular receptor subtype are important factors.
For example, the adrenergic receptor subtype associated with bronchiolar smooth muscle is principally β2 and, in accord with the activity of isoproterenol (Isuprel) at β2 receptors bronchodilation is the likely consequence of isoproterenol administration.
By contrast, the catecholamine norepinephrine would not be likely to cause this bronchiolar dilation because the agent exhibits relatively limited activity at β2 receptors.
Alpha receptors (α-receptors) are notably present in cutaneous vascular beds; at that site, both norepinephrine and epinephrine promote vasoconstriction.
Since isoproterenol has limited activity at α-receptors, isoproterenol will exhibit limited pharmacological effects in cutaneous vascular beds.
In the case of skeletal muscle vasculature, both α- and β-receptors are present with α-receptor activation mediating vasoconstriction and β-receptors promoting vasodilation.
The net effect may be vasodilation (β2-receptor-mediated) since the β2 adrenergic receptors are activated at lower catecholamine concentrations.
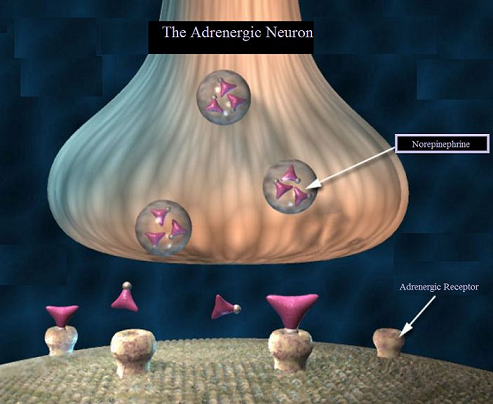
Agents which produce physiological effects similar to endogenous agents, such as the catecholamine epinephrine or norepinephrine, are often described as "sympathomimetics."
These agents mimic the action of substances released by the sympathetic part of the autonomic nervous system.
For example, suppose a sympathomimetic agent is injected and we are going to analyze its effects on blood pressure.
An important characteristic of the autonomic nervous system is that one must consider not only direct action of the agent on its target receptors but also indirect, compensatory effects which tend to restore the original homeostatic condition.
In our example, the sympathomimetic agent increases blood pressure through activation of α-receptors localized in the vascular bed, causing vasoconstriction and blood pressure elevation.
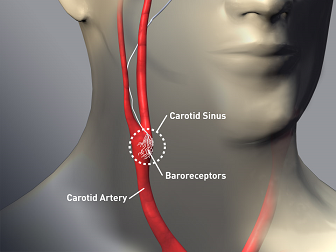
This increase in blood pressure is detected by receptors, e.g. baroreceptors, localized in carotid and aortic bodies.
Detection of blood pressure elevation induces a compensatory reflex which are one hand decreases central sympathetic outflow and also increases parasympathetic outflow.
Increased cardiac parasympathetic tone reduces heart rate and a reduction in sympathetic tone would likely reduce cardiac contractility.
As a result of these actions, the blood pressure would tend to return to lower levels, perhaps levels similar to those prior to sympathomimetic injection.
|
|
|
|
|
Adrenergic Drug Classification4
|
"Autonomic Nervous System (Pharmacology, Receptors and Physiology" |
|
|
|
Sympathomimetic drugs, as well as naturally occurring catecholamines, can be classified in terms of direct acting agents, agents which act almost exclusively by indirect physiological mechanisms and those which combine both indirect and direct actions.
Some agents exhibit much higher affinities for particular receptor subtype, so much so that they are defined as almost exclusively associated with that adrenergic subtype.
One of the best examples for α-receptors is phenylephrine (Neo-Synephrine™), which exhibits high affinity for the α1-receptor (α-subtype 1 receptor) subtype.4
|
|
|
Terbutaline (Brethine™) is an example among β-receptors as this agent shows relatively high specificity for the β2 -adrenergic receptor.4
|
|
|
The idea of relative specificity for drugs and receptors is based on their thermodynamic affinity (Kd).
KD = ([Drug] [Receptor]) / [Drug-Receptor Complex); D + R ↔ D-R
An agent may have a relatively high affinity (i.e. low Kd) for receptor subtype at a given drug concentration; however, at a 10 fold higher drug concentration the drug may interact with other receptors as well.
Therefore, often the selectivity of the drug for a particular receptor or receptor subtype is closely tied to its concentration at the receptor.
This idea also provides one explanation for side effects. Side effects may more likely occur at higher drug concentrations where drug-receptor interactions occur not only at the desired receptor site but also at other receptor sites.
Epinephrine is an example of an agent which acts both on α-receptors and β-receptors; specifically, epinephrine binds to α1-receptors, α2-receptors, as well as β1-receptors, β2-receptors, and β3-receptors.4
|
|
|
By contrast, norepinephrine exhibits affinity principally for α1-receptors, α2-receptors, and β1-receptors.
|
|
|
Indirect acting drugs may not of themselves have pharmacological activity that may increase the concentration of the endogenous agents norepinephrine and/or epinephrine.4
Mechanisms that may account for these effects include: 4
(1) promoting release or displacement of the endogenous catecholamine from storage sites
(2) inhibiting reuptake of the catecholamine, e.g. norepinephrine away from its receptor sites of action and into sympathetic neurons, and
(3) inhibition of enzymes which metabolized the catecholamine, thus reducing the concentration of active drug. Examples of such inhibitors include pargyline, an inhibitor of monoamine oxidase (MAO) and entacapone which inhibits catechol-O-methytransferase (COMT).
|
|
|
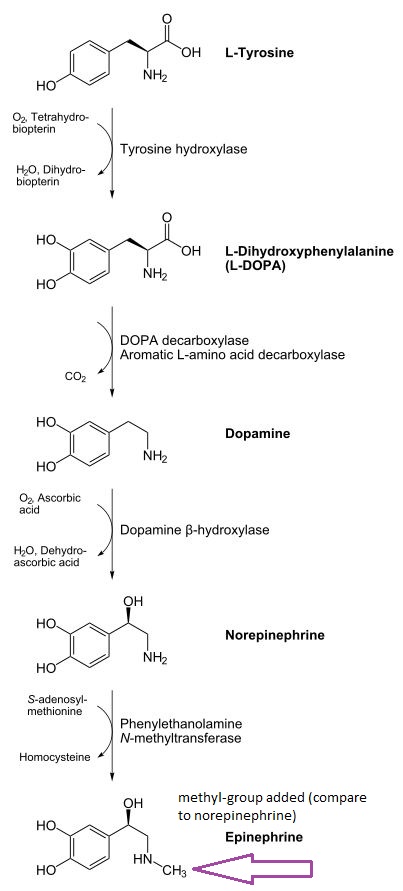
COMT (catechol-O-methyltransferase) is an enzyme that catalyzes a methyl group transfer from the methyl group donor, S-adenosyl-L-methionine. 6
For example, the anti-Parkinson's drug DOPA (e.g. l-dopa, levo-dopa) is metabolized to 3-O-methylDOPA.
Administration of levodopa by the oral route of administration results in about 99% of the drug inactivated by catabolism; furthermore, this inactivation is due to the action of the enzyme aromatic L-amino acid decarboxylase (AAD), converting levodopa peripherally (outside the brain) to dopamine.
Dopamine at these levels are associated with side effects such as nausea and hypotention; however, this conversion can be attenuated by the use of an AAD inhibitor, notably carbidopa.
This is where the issue of COMT inhibition comes in.
Carbidopa reduces extent of dopamine formation from levodopa but increases the fraction of levodopa methylated by COMT.
Entacapone (Comtan™) is an example of a catechol-O-methyltransferase inhibitor which blocks peripheral conversion of levodopa to 3-O-methylDOPA. With this levodopa degradation pathway inhibited, a half-life of levodopa is extended and more drug is available to enter the brain, its therapeutic side of action.
|
|
|
The other COMT inhibitor available in the United States is tolcapone (Tasmar™). These agents appear effective in decreasing the clinical symptom, described as "wearing off" in patients whose Parkinson's disease is managed with levodopa/carbidopa. 6
Some indirect acting agents may also directly activate receptors. Sometimes these agents are described as mixed-acting sympathomimetic agents and include ephedrine and dopamine.
|
|
|
"Catecholamine Biosynthesis Pathway" |
|
|
|
Direct vs. Indirect acting sympathomimetics4
Direct-acting sympathomimetic drugs are so named because they interact with the adrenergic receptor; therefore, the effects of these agents are not affected by changes in presynaptic catecholamine concentrations.
Some drugs deplete presynaptic endings of catecholamines which results in reduced sympathetic action. Direct-acting drugs/endogenous agents are not sensitive to these sorts of effects.
Examples of drugs which deplete presynaptic adrenergic terminals of their catecholamine stores include reserpine and guanethidine.
|
|
|
|
|
|
Although guanethidine exhibits pharmacological effects by several mechanisms, one mechanism is based on its accumulation within presynaptic catecholamine-storage vesicles and the subsequent depletion of norepinephrine within these terminals.
Over time there may be an increased sensitivity at the synaptic junction to circulating catecholamines; this effect resembles a type of "denervation hypersensitivity."8
Although the effect of direct-acting agents would not be influenced, at least not immediately, by presynaptic catecholamine depletion, indirect-acting agents would be quite sensitive to these effects.
Examples of indirect-acting drugs whose effects would be greatly diminished or eliminated by reserpine or guanethidine pretreatment include tyramine and amphetamine.
|
|
|
|
|
|
Mixed-acting drugs, the example used earlier was ephedrine, would have their action somewhat diminished but not eliminated by reserpine or guanethidine pretreatment.
|
|
|
A summary of direct-acting, mixed-acting, and indirect-acting adrenergic agonists is outlined:4
Direct-Acting Drugs
Receptor-type Selective Examples:
α1-Phenylephrine
α2-Clonidine
β1-Dobutamine
β2-Terbutaline
Receptor-type Non-Selective Examples:
α1,α2-Oxymetazoline
β1,β2-Isoproterenol
α1,α2-β1,β2-Epinephrine
α1,α2-β1,-Norepinephrine
Indirect-Acting Drugs
Releasing Agents:
Tyramine
Amphetamine
Uptake Inhibitor
Cocaine, which is not a sympathetic agent, but produces sympathomimetic effects due to reduced norepinephrine reuptake.
Uptake of norepinephrine is a principal mechanism for termination of physiological action.
Monoamine oxidase/ Catechol-O-methyltransferase (MAO/COMT) enzyme inhibitors
Pargyline (MAO inhibitor)
Entacapone (COMT inhibitor)
Mixed-Acting Drug Example
Ephedrine (direct activity at α1,α2-β1,β2 receptors AND also promotes catecholamine release from presynaptic storage sites)
|
Drug |
α |
β1 |
β2 |
Mechanism of action |
Peripheral resistance |
Renal blood flow |
Mean arterial pressure |
CNS stimulation |
|
Epinephrine |
|
|
|
Direct |
+/- |
|
|
Yes |
|
Norepinephrine (Levophed) |
|
|
0 |
Direct |
|
|
|
No |
|
Dopamine (Intropin) |
|
|
|
Direct |
|
|
|
No |
|
Isoproterenol (Isuprel) |
0 |
|
|
Direct |
|
|
+/- |
Yes |
|
Dobutamine (Dobutrex) |
0 |
|
0 |
Direct |
NC |
|
|
|
|
Ephedrine |
|
|
|
Direct+Indirect |
|
|
|
Yes |
|
Mephentermine (Wyamine) |
|
|
|
Direct+Indirect |
|
|
|
Yes |
|
Amphetamines |
|
|
|
Indirect |
|
|
|
Yes |
|
Metaraminol (Aramine) |
|
|
|
Indirect+direct |
|
|
|
No |
|
Phenylephrine (Neo-synephrine) |
|
0 |
0 |
Direct |
|
|
|
No |
-increased effect;
-decreased effect
adapted from: Table 12-1 Stoelting, R.K., "Pharmacokinetics and Pharmacodynamics of Injected and Inhaled Drugs", in Pharmacology and Physiology in Anesthetic Practice, Lippincott-Raven Publishers, 1999, p. 2602
|
|
|
|
|
|
|
|
|
This Web-based pharmacology and disease-based integrated teaching site is based on reference materials, that are believed reliable and consistent with standards accepted at the time of development. Possibility of human error and on-going research and development in medical sciences do not allow assurance that the information contained herein is in every respect accurate or complete. Users should confirm the information contained herein with other sources. This site should only be considered as a teaching aid for undergraduate and graduate biomedical education and is intended only as a teaching site. Information contained here should not be used for patient management and should not be used as a substitute for consultation with practicing medical professionals. Users of this website should check the product information sheet included in the package of any drug they plan to administer to be certain that the information contained in this site is accurate and that changes have not been made in the recommended dose or in the contraindications for administration. Advertisements that appear on this site are not reviewed for content accuracy and it is the responsibility of users of this website to make individual assessments concerning this information. Medical or other information thus obtained should not be used as a substitute for consultation with practicing medical or scientific or other professionals. |